

Norrie disease (ND) is an X linked genetic disorder causing bilateral blindness in early infancy because of severely dysplastic retinas. The major disease phenotype is a retrolental mass caused by undifferentiated, proliferated vitreous and retina, accompanied by maldeveloped anterior segment, leading to atrophy of the eyeball. A certain number of patients show psychomotor retardation or hearing impairment as part of a multisystem disorder.1 After the identification of the gene for ND, various types of pathological mutations have been documented in different ethnic groups.2,3 Phenotype-genotype evaluation has clarified that a mutation in the ND gene may also be responsible for a separate clinical entity, exudative vitreoretinopathy, retinopathy of prematurity, or unclassified retinal dysplasia.4,5 The clinical distinction between sporadic ND and bilateral persistent hyperplastic primary vitreous (PHPV) can be difficult.6,7 We report a novel truncating mutation of the ND gene in a Japanese family, in whom the proband was initially diagnosed with a sporadic form of PHPV.
Case report
The proband (patient 1) was the first son of non-consanguineous parents. He was born in 1993 with full term uneventful delivery and his mother noticed bilateral leucocoria 2 weeks after birth. An ophthalmologist suggested a severe case of PHPV in both eyes, and the patient underwent vitreous surgery on the right eye at 4 months of age because of the haemorrhage behind the lens. The patient was referred to our hospital at 15 months of age for further clinical distinction and prognostic counselling of his ocular conditions. On examination, he had normal psychomotor development and normal hearing ability. Pupils were not responsive to light and the visual acuity was no light perception in both eyes. The corneal diameter was 4 mm right eye and 9 mm left eye and band-shaped keratopathy was present in both eyes. A vascularised retrolental fibrous mass was barely visible behind the hazy lens accompanied by posterior synechiae in each eye. Ultrasonography showed a funnel-shaped retrolental mass in each eye. Scotopic electroretinograms were non-recordable. Visual evoked potential measurements revealed tentative P100 wave forms in both eyes. Eyeballs showed a gradual shrinkage in subsequent follow up studies to 7 years of age.
The parents and their second son were healthy with normal vision. The other family members had lived long in the Osaka area, Japan. According to the statements from the parents, the family members had normal vision. At this phase, patient 1 was thus diagnosed as a sporadic, severe form of bilateral PHPV.
In 2000, the third son (patient 2) was born at full term with uneventful delivery. The parents noticed bilateral leucocoria at birth. On examination at 6 days of age, the pupils were not responsive to light. Corneal diameters were 9 mm in both eyes. Each eye had a clear media but showed a vascularised retrolental whitish-yellow mass with ectropion uveas and posterior synechiae. Ultrasonography showed a funnel-shaped retrolental mass in each eye with axial lengths of 16 mm in both eyes. Magnetic resonance imaging revealed a more detailed feature of the retrolental masses (Fig 1). Results of electroretinograms and visual evoked potential measurements were similar to those of patient 1. During the subsequent follow up period, the eyeballs of patient 2 showed a gradual shrinkage and he remained unremarkable in neurological or otological studies.
Figure 1.
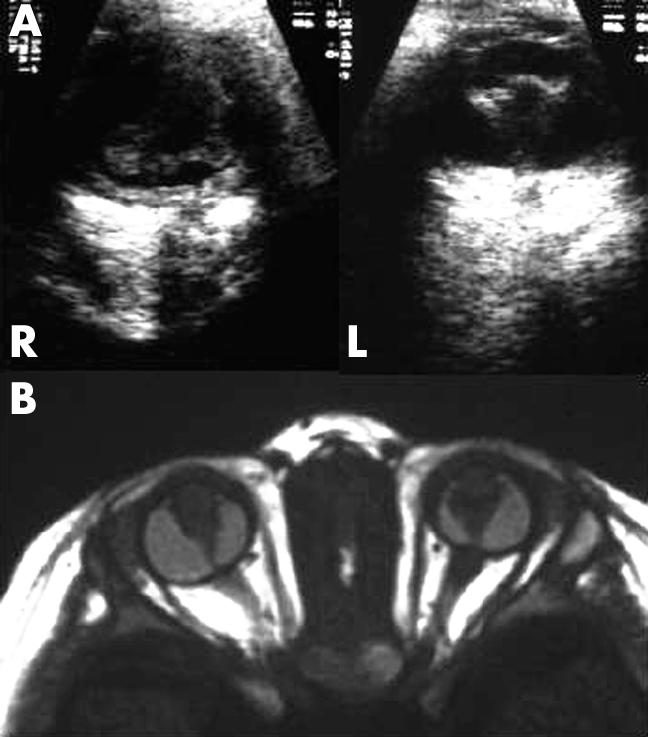
Ocular findings in patient 2. (A) R = right eye; L = left eye. Ultrasonography suggests a funnel-shaped retrolental mass in each eye with shortened axial lengths. (B) T1 weighted magnetic resonance imaging clarifies microphthalmic eyes, retrolental masses, abnormal lenses, and enhancement in the anterior chambers due to elongated ciliary processes.
We obtained peripheral blood with informed consent and analysed DNA samples of patient 2, his mother, and 100 normal controls, for mutations in the ND gene. One female and three male sporadic patients with bilateral PHPV were examined simultaneously as pathological controls. Patient 1 was not accessible for the molecular genetic examination. The coding exons of the ND gene were amplified according to the published data.8 Sense and antisense nucleotide sequences of the amplicons were directly determined by dye terminator autocycle sequencing. Patient 2 had a hemizygous mutation at Tyr 44 of the ND gene—namely, an insertion of adenine (TAT to TAAT) that creates a premature terminator (Ochre) to predict elimination of the subsequent amino acids of the ND protein (Fig 2). His mother had the normal and mutant types of the gene, which was expected for heterozygotes of the disease. On the other hand, four patients with bilateral PHPV and 100 normal controls showed only wild type sequences of the relevant gene.
Figure 2.

Sequencing results of the ND gene. Patient 2 has a hemizygous mutation at codon 44 of the ND gene—namely, an insertion of adenine (TAT to TAAT, arrow) that creates a premature terminator (Ochre) to predict elimination of the subsequent amino acid of the ND protein.
Comment
In Japanese families, ND gene mutations have been identified at the initiation codon (Met to Val) and codon 95 (Cys to Arg).9 This Japanese family shows a novel nonsense mutation (Tyr44stop) of the ND gene in a manner expected for an X linked genetic disease. This is the fourth Japanese family with ND in whom mutations of the ND gene were identified. We are unaware of previous reports of this mutation and could find no reference to it in a computerised search utilising Online Mendelian Inheritance in Man (OMIM) or the Cardiff Human Gene Mutation Database (HGMD).2,3 Among 60 types of ND gene mutations in the literature, seven nonsense mutations are located at Ser 29, Ser 57, Ser 73, Arg 109, Cys 110, Cys 126, and Cys 128, with typical clinical features of ND. In the manner similar to these mutations, Tyr44stop in the present report presumably eliminates subsequent 90 amino acid peptide when the gene is expressed. It is therefore likely that the present mutation has a strong effect leading to the ND phenotype.
The proband (patient 1) in the present family had been diagnosed as a sporadic form of bilateral PHPV until the third male offspring (patient 2) was born blind. Magnetic resonance imaging may be superior in demonstrating the retrolental masses to computed tomography imaging or ultrasonography.10 Magnetic resonance imaging in patient 2 clarified a detailed feature of the intraocular abnormalities including retrolental masses, abnormal lenses, elongated ciliary processes, and microphthalmic eyes. This type of morphological appearance would not be inconsistent with PHPV. It is clinically difficult to distinguish ND from PHPV with undefined aetiology especially in sporadic cases and the diagnostic confusion could be overcome by molecular genetic assessments. ND gene analysis has contributed the clinical diagnosis of a simplex patient with either ND or PHPV.6 However, two sporadic patients with bilateral PHPV were negative for mutations in the ND gene.7 In our series of sporadic patients with bilateral PHPV, we could not find any abnormalities in the ND gene. Thus, the frequency of ND gene mutation is very low in PHPV populations. The molecular genetic assessment of the ND gene enables us to make early diagnosis and give useful information for the genetic counselling.
Acknowledgments
This work was supported by grants in aid for scientific research (14571681) from the Japanese Ministry of Education, Science and Culture.
References
- 1.Warburg M. Norrie’s disease: a new hereditary bilateral pseudotumour of the retina. Acta Ophthalmol (Copenh) 1961;39:757–72. [Google Scholar]
- 2.Online Mendelian Inheritance in Man (OMIM). http://www.ncbi.nlm.nih.gov/Omim/
- 3.The Human Gene Mutation Database Cardiff. http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html
- 4.Chen ZY, Battinelli EM, Fielder A, et al. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat Genet 1993;5:180–3. [DOI] [PubMed] [Google Scholar]
- 5.Shastry BS, Pendergast SD, Hartzer MK, et al. Identification of missense mutations in the Norrie disease gene associated with advanced retinopathy of prematurity. Arch Ophthalmol 1997;115:651–5. [DOI] [PubMed] [Google Scholar]
- 6.Chynn EW, Walton DS, Hahn LB, et al. Norrie disease: diagnosis of a simplex case by DNA analysis. Arch Ophthalmol 1996;114:1136–8. [DOI] [PubMed] [Google Scholar]
- 7.Pendergast SD, Trese MT, Liu X, et al. Study of the Norrie disease gene in 2 patients with bilateral persistent hyperplastic primary vitreous. Arch Ophthalmol 1998;116:381–2. [PubMed] [Google Scholar]
- 8.Meindl A, Berger W, Meitinger T, et al. Norrie disease is caused by mutations in an extracellular protein resembling C-terminal globular domain of mucins. Nat Genet 1992;2:139–43. [DOI] [PubMed] [Google Scholar]
- 9.Isashiki Y, Ohba N, Yanagita T, et al. Mutations in the Norrie disease gene: a new mutation in a Japanese family. Br J Ophthalmol 1995;79:703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Edward DP, Mafee MF, Garcia-Valenzuela FE, et al. Coats’ disease and persistent hyperplastic primary vitreus: role of MR imaging and CT. Radiol Clin North Am 1998;36:1119–31. [DOI] [PubMed] [Google Scholar]