

Abstract
Aims: To establish a clinical, histopathological, and genetic diagnosis in two unrelated British families with Avellino corneal dystrophy (ACD).
Methods: Genomic DNA was extracted from peripheral blood leucocytes of all members participating in the study. Exons 4 and 12 of the human transforming growth factor β induced (BIGH3) gene were amplified by polymerase chain reaction. The mutation and polymorphism were identified by direct sequencing and restriction digest analysis. A review of the patients’ clinical symptoms and signs was undertaken and a histopathological study on corneal specimen obtained from the proband of one family after keratoplasty was performed.
Results: A heterozygous G to A transition at the second nucleotide position of codon 124 of BIGH3 gene was detected in all affected members of both families. This mutation changes an arginine residue to a histidine. The clinical diagnosis for ACD was more evident with advancing age. Histopathological study revealed granular deposits in the anterior stroma and occasional positive Congo red areas of amyloid deposition in the mid to deep stroma typical of ACD.
Conclusions: This is the first report of ACD families in the United Kingdom and, furthermore, of BIGH3 gene mutation in British patients with this rare type of corneal dystrophy. The results indicate that BIGH3 gene screening along with clinical and histopathological examinations is essential for the diagnosis and clinical management of corneal dystrophies.
Keywords: Avellino corneal dystrophy, British families
Avellino corneal dystrophy (ACD; OMIM 121900) is a variant of granular corneal dystrophy Groenouw type 1 (CDGG1; OMIM 12100) in which both Groenouw-like and lattice-like changes co-exist in the same cornea.1 The condition has been named Avellino, after the Italian province near Naples where the first affected families originated.1 However, the condition has also been reported in Germany,2 Ireland,3 Europe,4 Japan,5–8 and France.9
The earliest clinical evidence of ACD is the development of small, discrete and sharply demarcated granular deposits in the subepithelial and anterior stromal layers of the cornea. As the condition progresses the deposits increase in size and number, and lattice lesions develop in the mid to posterior corneal stroma.10 These lattice lesions are larger, denser, whiter, and more spiculated than those of lattice corneal dystrophy type I (CDLI; OMIM 122000).11 Stromal haze is the last clinical sign to emerge; thus, all patients with stromal haze had both granular and lattice lesions, representing the most advanced form of the disease.10
Histologically, granular deposits that stained with Masson’s trichrome and Congo red positive fusiform deposits of amyloid are found in the corneal stroma.2,12–14
ACD, CDGGI, CDLI, and Reis-Bücklers corneal dystrophy (RBCD; OMIM 121900), have been found to be due to different missense mutations within a human transforming growth factor β induced (BIGH3; OMIM 601692) gene on chromosome 5q31.4 ACD has been reported to be caused by heterozygous R124H mutation in the BIGH3 gene.4
Here, two unrelated British families have been studied clinically, histopathologically and genetically. Our results revealed a heterozygous G to A transition at the second nucleotide position of codon 124 in the BIGH3 gene in all affected members of both families. Clinically the diagnosis of ACD was more evident in the older individuals rather than younger cases and the histopathological diagnosis of one case who underwent keratoplasty was typical for ACD. These results further highlight on the importance of BIGH3 gene screening to establish a precise diagnosis of corneal dystrophies.
METHODS
Case reports
The study had the approval of Moorfields Eye Hospital local research ethics committee and conformed to the tenets of the Declaration of Helsinki. Informed consent from all participants was obtained for clinical and molecular genetic study. The pedigrees are shown in Figure 1.
Figure 1.

Pedigrees of the families participating in the study. Open and closed symbols indicate unaffected and affected individuals, respectively, arrows indicate the proband in each family, and asterisks indicate individuals examined.
Family A
Case II-1 (the proband) was first examined at the cornea and external disease clinic in 1990 at the age of 72 years, when she complained of visual disturbance. The patient’s best corrected visual acuity at the initial examination was 6/60 in the right eye and 6/18 in the left eye. Slit lamp biomicroscopic examination showed discrete dot opacities with interspersed thick lattice lines in between and the condition was diagnosed as an atypical form of granular corneal dystrophy (Fig 2). The patient also had cataract but no other ocular abnormalities were present. Extracapsular cataract extraction and intracapsular lens implantation were performed in the right eye in 1990, where right eye visual acuity was improved to 6/36 unaided. Penetrating keratoplasty in the right eye was then performed in 1991 which further improved her visual acuity to 6/18 unaided.
Figure 2.
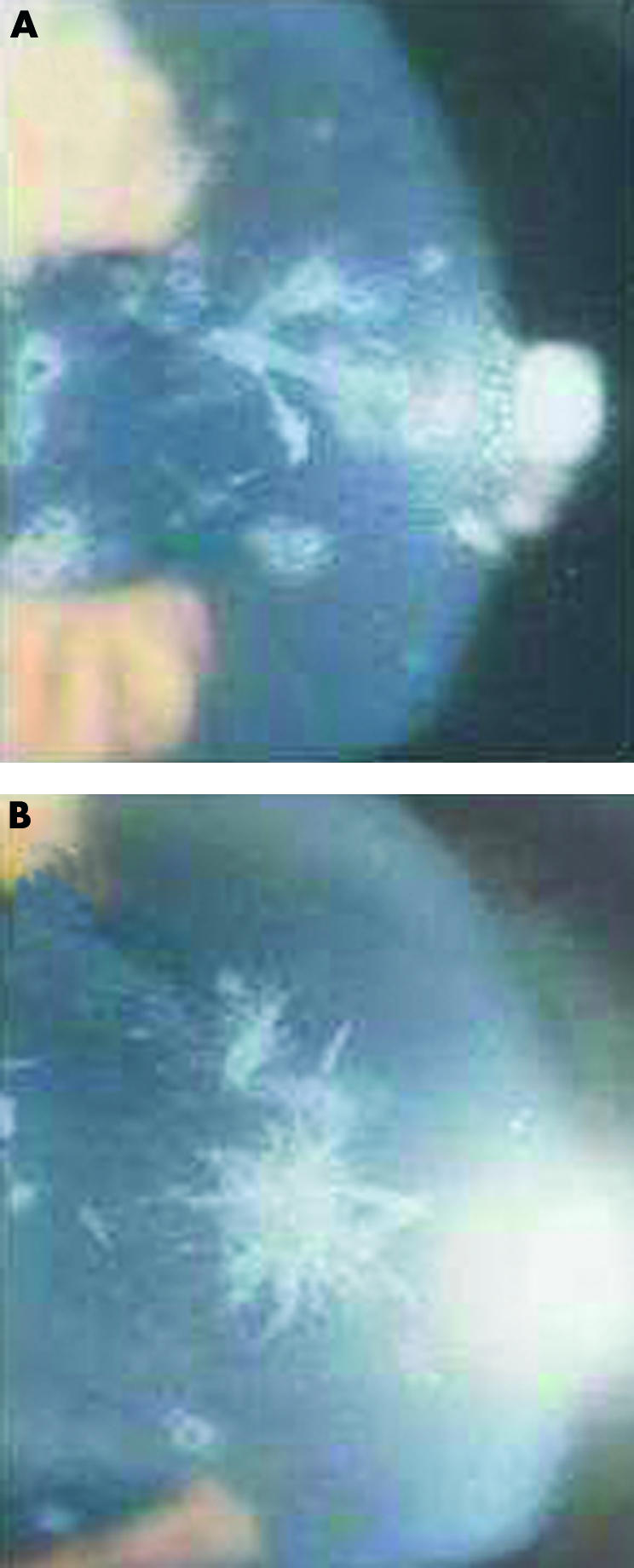
Slit lamp photographs of ACD in the upper (A) and lower (B) corneas of the proband from family A showing fusiform, branching, and white breadcrumb deposits.
Histopathological examination of her corneal specimen showed an amorphous eosinophilic deposit in the anterior stroma, which stained positively with Masson’s trichrome stain and occasional areas of amyloid deposition in the deep stroma, which stained positively with Congo red stain (Fig 3). These criteria were typical of ACD.
Figure 3.

Light microscopic study of corneal specimen from the proband of family A. (A) Subepithelial fusiform and anterior stromal deposits stained pink with Congo red (×10). (B) Focal accumulation of hyaline stained bright red with Massone’s trichrome; subepithelial hyaline material and connective tissue (×20). (C) Characteristic birefringence seen with polarised light in amyloid deposits (×20).
Case III-3 (the proband’s son) was referred to the same clinic in 1996 at the age of 38 years, when he complained of bilateral decrease in visual acuity. He had been diagnosed as having a corneal dystrophy when he attended the casualty department for removal of a foreign body in 1980. The patient’s unaided visual acuity was 6/9 in either eye improving to 6/6 with pinhole. Slit lamp examination revealed discrete granular deposits in the anterior stromal layer and star-like deposits which are typical of ACD. In 1994 the patient’s visual acuity deteriorated and right excimer laser phototherapeutic keratectomy had been performed, which improved his vision.
Case III-1 (the proband’s daughter) was a 25 year old and the slit lamp examination of her cornea revealed large number of central granules, but no lattice changes were observed. The patient’s unaided visual acuity was 6/9 in both eyes and she required no intervention.
Case II-5 (the proband’s sister) was completely free of corneal intervention as determined by slit lamp biomicroscopic examination of her corneas.
Family B
The second family consisted of 90 year old proband (case I-1) and 63 year old son (case II-1). Slit lamp examination of their corneas showed a typical picture of ACD. They had no history of keratoplasty. Examination of the proband’s daughter (case II-2) revealed that she was completely free of corneal disease.
All members of both families were born in England and both families’ origin has been traced to the United Kingdom. They have no relatives of Italian origin, making them the first British families to be reported with ACD.
Mutation detection
Genomic DNA was extracted from 10 ml blood samples obtained from all subjects involved in the study according to the standard protocol. Exons 4 and 12 of BIGH3 gene were amplified by polymerase chain reaction (PCR) using the following primer pairs (exon 4, 5′-tccctccttctgtcttctgc -3′/5′-agactcccattcatcatgcc-3′; exon 12, 5′-tcaatccttgatgtgccaac-3′/5′-aaaatacctctcagcgtggtg-3′). Each PCR reaction was carried out in a 50 μl reaction mixture containing genomic DNA (200 ng), primers (0.4 μM each), MgCl2 (1.5 mM), dNTPs (0.2 mM), 1X PCR buffer (containing 10 mM TRIS-HCl, pH 8.3, 50 mM KCl, and 0.1% gelatin), and Taq polymerase (0.5 U; Bioline). Amplification reactions were performed using the following conditions (3 minutes of denaturation at 94°C followed by 35 cycles of denaturation at 94°C for 1 minute, annealing at 62°C for 1 minute, extension at 72°C for 1 minute, and further extension step at 72°C for 5 minutes).
For direct sequencing, PCR products were purified using the Qiaquik PCR purification kit (Qiagen) and sequenced using an automatic fluorescent DNA sequencer (ABI Prism 373A, Perkin Elmer, Foster City, CA, USA) following manufacturers instructions. Nucleotide sequences for the coding regions were compared with the nucleotide sequence of the published BIGH3 human cDNA.15
Co-segregation study on the family members was performed using restriction enzyme digest analysis to confirm the mutation detected by sequencing. Exon 4 primer pairs were used to PCR amplify a 310 bp product as described above, which was subsequently digested overnight at 30°C with 1 unit of CSPI enzyme (Promega) and analysed on 3% agarose gel. The recognition sequence for the enzyme is 5 …[/]CGG[_](A/T)CCG[/ …]3, which is abolished by the mutation.
RESULTS
Direct sequencing analysis of BIGH3 exon 4 in the probands from both families participating in this study revealed a single heterozygous base pair transition at nucleotide position 418 (G to A), which converts an arginine at codon 124 into a histidine shown in Figure 4. This mutation has previously been identified in ACD.4
Figure 4.
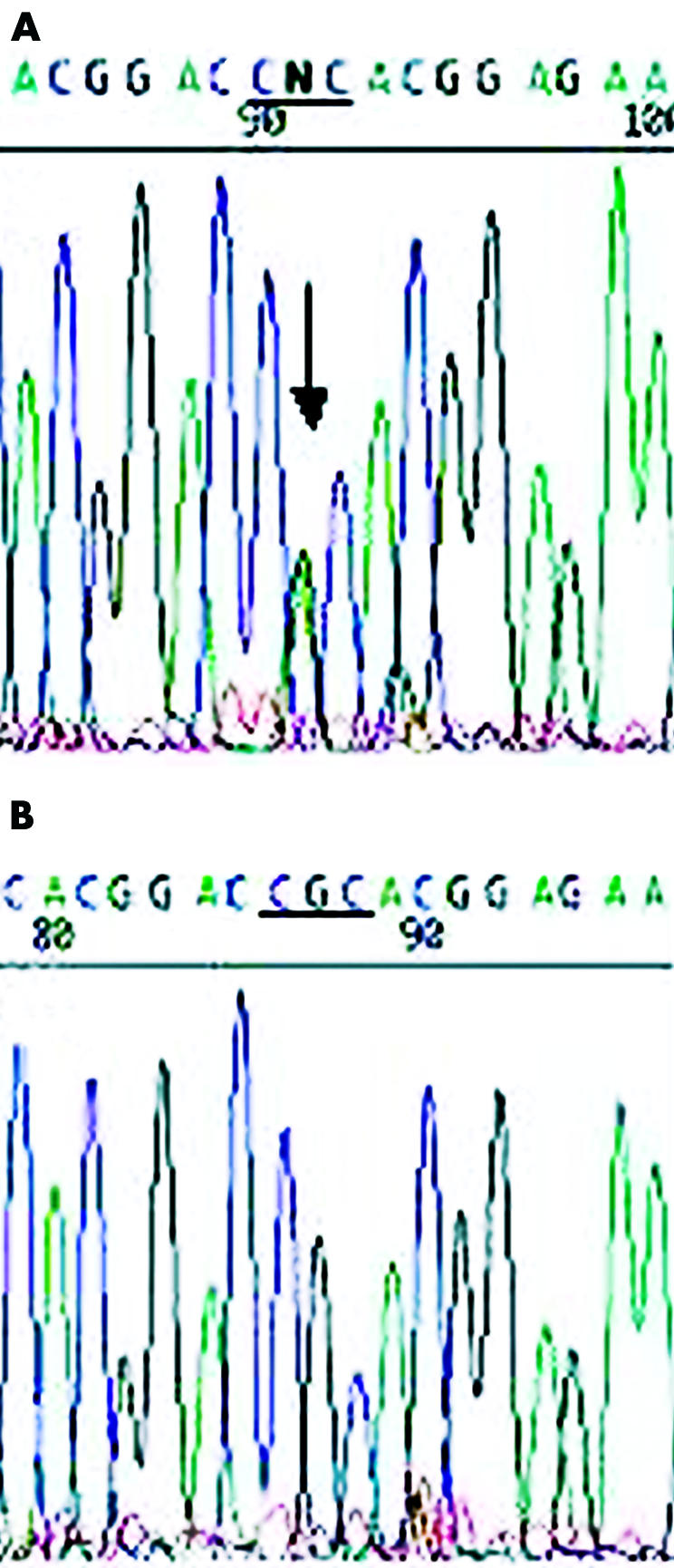
Electropherograms of the BIGH3 gene. (A) The proband of family A showing a heterozygous G to A transition at codon 124, (CGC→CAC, R124H). (B) The proband’s daughter from family A depicting normal sequence.
The unaffected members of both families did not have the mutation as determined by a cosegregation study using restriction analysis in which the mutation abolishes the recognition site of CSPI. The three affected members from family A and the two affected from family B showed one undigested product of the expected size (310 bp) for the mutant allele plus two restriction digest products (190 bp and 110 bp) from the normal allele. Unaffected members from both families revealed two restriction digest fragments (190 bp and 110 bp) as shown in Figure 5.
Figure 5.

Co-segregation study with CSPI restriction enzyme. Lane 1 is undigested PCR product (310 bp). The two affected individuals (lanes 2 and 3) from family B showing one undigested product of the expected size (310 bp) for the mutant allele plus two restriction digest products (190 and 110 bp) from the normal allele. Unaffected member (lane 4) showing two restriction digest fragments (190 and 110 bp). M is the φX174 RF DNA Hae III marker.
BIGH3 exon 12 sequencing revealed a polymorphism with T to C transition at nucleotide position 1620 in the proband from family B. This polymorphism does not change the amino acid sequence of the encoded protein, and it has also been reported previously.16,17
DISCUSSION
ACD is an autosomal dominant disorder that usually appears in the first or second decade of life.1 It consists of granular and lattice changes in the same eye and results from a specific mutation in the BIGH3 gene on chromosome 5q31.4
The majority of ACD families were found to trace their ancestry to the large region of Campania in Italy which contains cities of Naples, Avellino, Lioni, and Stio. However, many cases of ACD have been reported in patients of different origins, including German, Japanese, Irish, Swiss, and French.2–6,8,9
In this study we describe for the first time two pedigrees of ACD in the United Kingdom indicating that the condition may occur in any population.
The younger individual (case III-1 in family A) demonstrated predominantly granular stromal opacities and the appearance of lattice changes occurred gradually, starting later in life, and increasing with age. Thus, the dystrophic process progresses with age. This observation supports and extends those of previous reports.1,2,6,10,18
Molecular genetic analysis showed that all five patients in both families participating in this study, had heterozygous R124H BIGH3 gene mutation which is identical to the mutation reported previously for ACD.4 It appears that the unique phenotype of ACD is caused by this particular amino acid change. This fact is supported by the finding that different amino acid changes in the same position result in different phenotypes R124C results in LCDI,4 R124L results in the geographic form of RBCD,19 and R124S results in atypical form of CDGG1,20 and even the homozygous form of the same change (R124H) results in a severe variant of CDGG1 which is characterised by juvenile onset and confluent superficial discrete opacity.21 Thus the heterozygous R124H mutation of the BIGH3 gene is particularly linked to ACD phenotype. However, Stewart et al20 reported that the heterozygous R124H mutation of the BIGH3 gene resulted in an atypical form of CDGGI which was confirmed histopathologically by absence of amyloid deposits. Thus, genetic and histopathological evaluation are both essential for correct diagnosis of these dystrophies
In conclusion, this is the first reported BIGH3 gene mutation in ACD families from the United Kingdom. Our results confirm the importance of BIGH3 gene screening as a mandatory step for the diagnosis of ACD, particularly in the younger age group. This information will be useful for future genetic counselling, as well as gene therapy.
Acknowledgments
We would like to thank the families who participated in this study. A research grant from Tanta University Hospital supported this study.
REFERENCES
- 1.Folberg R, Alfonso E, Croxatto JO, et al. Clinically atypical granular corneal dystrophy with pathologic features of lattice-like amyloid deposits:a study of three families. Ophthalmology 1988;95:46–51. [DOI] [PubMed] [Google Scholar]
- 2.Rosenwasser GO, Sucheski BM, Rosa N, et al. Phenotype variation in combined granular-lattice corneal dystrophy. Arch Ophthalmol 1993;111:1546–52. [DOI] [PubMed] [Google Scholar]
- 3.Kennedy SMO, McNamara M, Hillery M, et al. Combined granular lattice dystrophy (Avellino corneal dystrophy). Br J Ophthalmol 1996;80:489–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in four 5q31 linked corneal dystrophies. Nat Genet 1997;15:247–51. [DOI] [PubMed] [Google Scholar]
- 5.Santo RM, Yamaguchi T, Kanai A, et al. Clinical and histopathologic features of corneal dystrophies in Japan.Ophthalmology 1995: 557–67. [DOI] [PubMed]
- 6.Konishi M, Yamada Y, Nakamura Y, et al. Granular-lattice (Avellino) corneal dystrophy in Japanese patients. Cornea 1997;16:635–8. [PubMed] [Google Scholar]
- 7.Konishi M, Yamada M, Nakamura Y, et al. Corneal dystrophy associated with R124H mutation in the Big-h3 gene. Cornea 1999;18:424–9. [DOI] [PubMed] [Google Scholar]
- 8.Akimune C, Watanabe H, Maeda N, et al. Corneal guttata associated with the corneal dystrophy resulting from a Big-h3 R124H mutation. Br J Ophthalmol 2000;84:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dighiero P, Ellies P, Valleix S, et al. Avellino corneal dystrophy. J Fr Ophtalmol 2000;23:735–7. [PubMed] [Google Scholar]
- 10.Holland EJ, Daya SM, Stone EM, et al. Avellino corneal dystrophy: clinical manifestations and natural history. Ophthalmology 1992;99:1564–8. [DOI] [PubMed] [Google Scholar]
- 11.Lucarelli MJ, Adamis AP. Avellino corneal dystrophy. Arch Ophthalmol 1994;112:418. [DOI] [PubMed] [Google Scholar]
- 12.Garner A. Histochemistry of corneal granular dystrophy. Br J Ophthalmol 1969;53:799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akiya S, Brown SI. Granular dystrophy of the cornea: characteristic electron microscopic lesion. Arch Ophthalmol 1970;84:179–92. [DOI] [PubMed] [Google Scholar]
- 14.Owens SL, Sugar J. Edward DP. Superficial granular corneal dystrophy with amyloid deposit. Arch Ophthalmol 1992;110:175–6. [DOI] [PubMed] [Google Scholar]
- 15.Skonier J, Neubauer M, Madisen L, et al. cDNA cloning and sequence analysis beta ig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-beta. DNA Cell Biol 1992;11:511–22. [DOI] [PubMed] [Google Scholar]
- 16.Korvatska E, Munier FL, Djemai A, et al. Mutation hot spots in 5q31-linked corneal dystrophies. Am J Hum Genet 1998;62:320–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmitt-Bernard CF, Guittard C, Arnaud B, et al. BIGH3 exon 14 mutations lead to intermediate type I/IIIA of lattice corneal dystrophies. Invest Ophthalmol Vis Sci 2000;41:1302–8. [PubMed] [Google Scholar]
- 18.Sassani J, Smith SG, Rabinowitz YS. Keratoconus and bilateral lattice-granular corneal dystrophies. Cornea 1992;11:343–50. [DOI] [PubMed] [Google Scholar]
- 19.Okada M, Yamamoto S, Tsujikawa M, et al. Two distinct kerato-epithelin mutations in Reis-Bucklers’ corneal dystrophy. Am J Ophthalmol 1998;126:535–42. [DOI] [PubMed] [Google Scholar]
- 20.Stewart HS, Ridgway AE, Dixon MJ, et al. Heterogeneity in granular corneal dystrophy:Identification of three causative mutations in the TGFB (BIGH3) gene-Lessons for corneal amyloidogenesis. Hum Mut 1999;14:126–32. [DOI] [PubMed] [Google Scholar]
- 21.Okada M, Yamamoto S, Inoue Y, et al. Severe corneal dystrophy phenotype caused by homozygous R124H keratoepithelin mutations. Invest Ophthalmol Vis Sci 1998;39:1947–53. [PubMed] [Google Scholar]