

Abstract
Behçet’s disease (BD) is characterised by recurrent episodes of orogenital aphthae, systemic vasculitis, and systemic and retinal venous thrombosis. An association between HLA-B51 and BD was first identified over 20 years ago, but recently identified gene associations implicate regions both within and without the MHC in the immunological events underlying the lesions in BD. These include allelic variants within the tumour necrosis factor gene region and within the MHC class I chain related gene region, the factor V Leiden mutation, which is associated with retinal vascular occlusion, and alleles of the intercellular adhesion molecule gene. No single causative gene for BD has emerged; the evidence indicates that the underlying immune events in BD are triggered by a microbial antigen and subsequently driven by genetic influences which control leucocyte behaviour and the coagulation pathways. Knowledge of these risk factors may permit a more accurate prognosis for a given patient, and identify new pathways for more targeted intervention than is currently available.
Keywords: Behçet’s disease, factor V Leiden, gene polymorphisms, retinal vascular occlusion, uvei
Behçet’s disease owes its name to the Turkish physician professor Huluci Behçet, who, in 1937, described the classic trisymptom complex of hypopyon, iritis, and orogenital aphthosis.1 The Greek physician Adamantiades had reported the disease 6 years earlier, accounting for the alternative eponym Adamantiades-Behcet disease.2 However, the first description probably predates both of these by 2500 years. In his Epidemion, book 3, case 7, Hippocrates of Kos (460–377 bc) describes an endemic disease in Asia Minor characterised by “aphthous ulcerations,” “defluxions about the genital parts,” “watery ophthalmies of a chronic character . . . which destroyed the sight of many persons,” and “large herpetic lesions.”3 No further reference to the disease is encountered in the medical literature until the early 1900s, when the classic trisymptom complex was again described in Europe. At first, this complex was considered to be a manifestation of syphilis, but in 1937 Behçet proposed a separate disease entity, which, by 1947, had gained widespread international recognition. In the absence of a diagnostic laboratory test, Behçet’s disease still remains a clinical diagnosis, and for the purposes of international research the diagnostic criteria proposed by the International Study Group in 1990 are now widely accepted (Table 1).4
Table 1.
International Study Group criteria for Behçet’s disease
| Criteria present | Description |
| Recurrent oral ulceration: | Minor aphthous, major aphthous, or herpetiform ulceration observed by physician or patient, which recurred at least 3 times in one 12 month period |
| Plus two of: | |
| Recurrent genital ulceration: | Aphthous ulceration or scarring, observed by physician or patient |
| Eye lesions: | Anterior uveitis, posterior uveitis, or cells in vitreous on slit lamp examination; or retinal vasculitis observed by ophthalmologist |
| Skin lesions: | Erythema nodosum observed by physician or patient, pseudofolliculitis, or papulopustular lesions; or acneiform nodules observed by physician in post-adolescent patients not on corticosteroids |
| Positive pathergy test: | Read by physician at 24–48 hours |
OCULAR DISEASE
Widespread organ involvement in Behçet’s disease is now well recognised (Fig 1), although the clinical course and degree of organ involvement is often similar in successive relapses for a given patient. Inflammatory eye disease occurs in approximately 70% of all patients, and although it typically occurs after the onset of oral aphthosis, the delay between the two may be as long as 14 years.5 Conversely, intraocular inflammation is the presenting feature in over 10% of patients,6 and in rare cases oral aphthosis may not occur at all.7 The majority of ocular cases eventually suffer bilateral disease, but in 6% of cases, disease remains uniocular.8 Features of acute intraocular inflammation and the sequelae of recurrent episodes are given in Table 2, and Figures 2–4. It is of great interest that, while oral and genital aphthosis occur frequently in Behçet’s disease, conjunctival ulcers are rarely encountered. Indeed, in a recent series of 1000 Iranian patients, conjunctival ulceration was described in only one individual,9 and in a recent Japanese study only four of 152 patients developed conjunctival ulceration over a 5 year period.10 This paradoxical sparing of the conjunctiva, and the complete absence of respiratory mucosal lesions, supports a role for infectious agents in the gastrointestinal tract in initiation and perpetuation of disease (see below).
Figure 1.

Spectrum of organ involvement in Behçet’s disease.
Table 2.
Ocular features of Behçet’s disease
| Features of active disease | Sequelae of recurrent inflammation |
| Anterior uveitis | Anterior and posterior synechiae |
| Hypopyon | Cataract |
| Inflammatory retinal vein occlusion | Cystoid macular oedema |
| Macular oedema | Glaucoma |
| Optic disc oedema | Neovascular glaucoma |
| Retinal infiltrates | Optic atrophy |
| Retinal detachment | Phthisis bulbi |
| Retinal and optic disc neovascularisation | Rubeosis iridis |
| Retinal vasculitis | Retinal vascular obliteration |
| Scleritis | |
| Vitritis |
Figure 2.
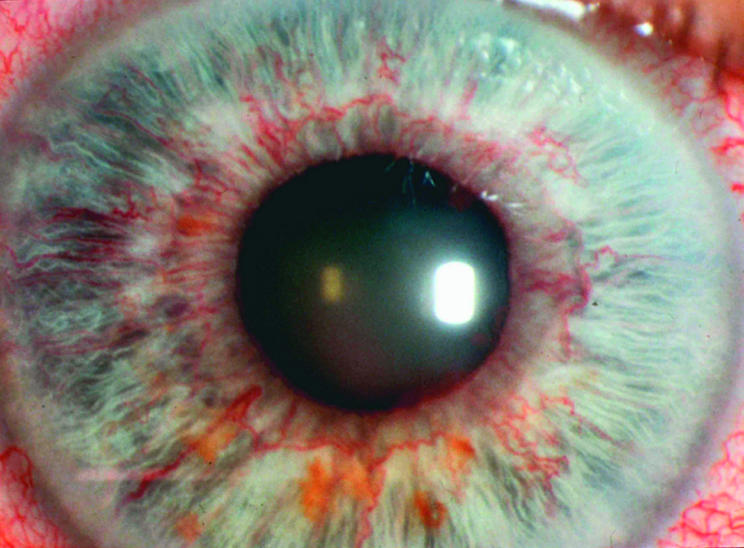
Anterior segment complications of retinal vein occlusion: retinal ischaemia with secondary rubeosis iridis.
Figure 3.

Inflammatory retinal vein occlusion with associated vitritis and retinal vasculitis before (A) and after (B) treatment with high dose oral steroid.
Figure 4.

(A) Acute branch retinal vein occlusion. (B) Total vascular obliteration and optic atrophy secondary to recurrent vascular occlusion.
The prognosis for vision in patients with recurrent posterior segment disease may be dismal, chiefly because of inflammatory retinal vein occlusions and macular oedema. In 1993, up to a quarter of eyes became blind.11 This dismal statistic has been reduced by modern immunosuppressive agents and more targeted immunomodulators, but up to 15% of ocular patients remain resistant to intervention.12 Cyclosporine, tacrolimus (FK 506), azathioprine, and anti-T cell monoclonal antibodies have all proved useful in controlling ocular disease in the short term, but their effect over the duration of active eye disease, which may be two to three decades, remains unproved.13–16 However, with increasing knowledge of the immune events that underlie Behçet’s disease, newer interventions have become more targeted. Activation of neutrophils and leucocyte production of both IL-8 and tumour necrosis factor (TNF) are examples of pathways that may targeted. Blockade of TNF with infliximab, a chimeric anti-TNF-α monoclonal antibody, is effective in diminishing the severity of systemic Behçet’s disease,17 and early reports on the effect on ocular disease are also promising.18,19
Recently, recombinant human interferon α-2a (rhIFNα-2a) has been employed in cases of refractory uveitis in Behçet’s disease with promising results.20,21 Kötter et al investigated the efficacy of subcutaneous rhIFNα-2a in 50 patients with highly resistant ocular Behçet’s disease, and reported a response in all but four patients.21 Equally significant is the finding that in 20 patients (40%), therapy could be discontinued after a mean period of 16.4 months without relapse over a mean observation period of 29.5 months. However, the mechanism of action of interferon in this context remains uncertain. IFN is known to inhibit γδ+ T cells,22 which have been implicated in the gastrointestinal lesions in Behçet’s disease, and in addition has antiviral properties, which may be relevant given a possible role for herpes simplex virus in disease. Both of these aetiological factors are discussed below. Finally, rhIFNα-2a may downregulate IL-8 production by activated neutrophils, although paradoxically neutrophil adhesion and phagocytosis are reported to be increased following IFNα-2a treatment in patients with Behçet’s disease.23 Further studies are therefore awaited to elucidate the mechanism of action of IFNα-2a in ocular Behçet’s disease.
PATHOLOGY
Behçet’s disease is a systemic perivasculitis, in which early neutrophil infiltration, endothelial cell swelling, and fibrinoid necrosis are well described.24 Significant neutrophil infiltration is seen in all early lesions, including mucocutaneous aphthae, the skin pathergy reaction (in which minor trauma triggers a rapid cutaneous inflammatory response), nodular cutaneous lesions, and ocular lesions.25,26 In addition, serum levels of neutrophil priming cytokines such as TNF, interleukin 1 β (IL-1β) and IL-8 are known to be raised in patients,27 and myeloperoxidase levels, generated by active neutrophils, are also raised.28 Whether neutrophil hyper-reactivity in Behçet’s disease reflects genetic influences or persistent activation by external priming agent(s) remains unresolved, although data from studies on familial Mediterranean fever (FMF), which shares features with Behçet’s disease and is encountered in similar geographic regions, suggest that neutrophil responses are indeed under genetic control, as discussed below.
BEHçET’S DISEASE—AN INFECTIVE AETIOLOGY?
Although a microbiological trigger may underlie the innate immune response at the mucosal surface in Behçet’s disease, the mechanism(s) for the vasculitis which follows (for example, inflammatory retinal vein occlusion) remain unknown, although they are not thought to have a direct microbial aetiology. The distribution of Behçet’s disease in countries between the Mediterranean and the Orient, and its virtual absence in indigenous Amerindians and among populations south of the equator is intriguing29 (Table 3). Early trade routes between the Mediterranean and the Orient (the Silk Roads) promoted both commerce and the spread of disease, as exemplified by the plague of Athens in 430 bc, and the Black Death (Yersinia pestis) in the 14th century, the cause of the worst pandemic of the Middle Ages.30 According to the Hippocratic description, Behçet’s disease may have been endemic in Asia Minor. No further descriptions emerge in the literature for two millennia, yet in the past 20 years a dramatic increase in reporting has occurred across southern Europe, Eurasia, and the Arabic world. This may reflect a reporting bias as the distinction between Behçet’s disease and both syphilis and tuberculosis became appreciated. Equally, the rise in incidence may be real, and may be the result of contributory environmental agent(s). Indeed, both Behçet and Adamantiadis suspected an infectious aetiology, and two areas of research now support a microbial role in the disease.
Table 3.
Association between latitude and prevalence of Behçet’s disease (adapted from Demirkesen et al25)
| Latitude (°N) | Country/ethnic group | Prevalence of Behçet’s disease per 105 |
| 25 | Saudi Arabia | 20 |
| 31 | Palestinian | >10 |
| 32 | Iran | 16.7 |
| 32 | Morocco | >15 |
| 36 | Japan | 13.5 |
| 39 | Portugal | 1.53 |
| 39 | Turkey | 100 |
| 42 | Italy | 2.5 |
| 52 | England | 0.5 |
| 56 | Scotland | 0.27 |
| 60 | Sweden | 1.18 |
| 62 | Amerindian | Not reported |
Firstly, skin and peripheral blood monocytes have been shown to exhibit a hypersensitivity response to certain Streptococcus sanguis antigens in patients but not in healthy controls.31 These bacterial antigens share amino acid sequences in common with one of the class of cell membrane proteins called heat shock proteins (HSP), which are expressed on cell membranes in response to physiological shock and microbial challenge. Several groups have demonstrated that both Streptococcus sanguis and the human HSP (especially the 60/65 kDa HSP) activate γδ T cells in patients but not controls.32,33 This observation has led to the suggestion that, following bacterial challenge, oral mucosal cells express HSPs which are antigenic and lead to antimucosal T cell cross reactivity in susceptible individuals (the molecular mimicry model). The observations that oral aphthae typically precede the onset of systemic disease by months or years,34 and frequently herald disease relapses, provide circumstantial support for this model. In addition, prophylactic penicillin has also been reported to reduce the frequency and duration of mucocutaneous symptoms of disease in a Turkish population.35 However, Behçet’s disease is rare, streptococcal species are common oral fauna, and HSP are ubiquitously expressed by stressed cells. This model would require an escape of T cell clonal deletion (or positive selection) in the thymus in Behçet’s disease or, alternatively, irregular leucocyte behaviour at the mucosal surface in patients, which may itself be influenced by certain HLA types. This second scenario is discussed at greater length later.
Behçet himself had proposed an aetiological role for herpes simplex virus (HSV) in the disease. Serum antibodies to HSV-1 and circulating immune complexes with HSV-1 are both reported to be raised in patients.36 Secondly, an animal model for Behçet’s disease was described in 1997, in which 30% of HSV-1 innoculated mice were reported to exhibit Behçet’s disease-like symptoms, which included skin, tongue, gastrointestinal and genital ulcers, and ocular inflammation and arthritis.37 Although HSV-1 immunity is common in the general population, the possibility exists that certain genetic haplotypes may influence the immune response of an individual to HSV-1 infection at the gastrointestinal mucosal surface. However, until such evidence arises, any role for HSV-1 in the pathogenesis of Behçet’s disease remains speculative.
HERITABLE RISK FACTORS FOR BEHçET’S DISEASE
Family studies
Behçet’s disease is not a monogenic disorder, and no clear Mendelian pattern of inheritance has yet emerged. Nevertheless, a number of features point to a genetic aetiology. The unusual geographical distribution of disease and its close association with the major histocompatibility complex (MHC) allele HLA-B51 (see below) are perhaps the strongest indicators that certain gene(s) are either directly responsible for disease, or promote indirectly the characteristic underlying inflammatory changes. Furthermore, familial aggregation in Behçet’s disease is well recognised. Recently, Gül et al reported the sibling recurrence risk ratio (λs) among patients in Turkey.38. λs is an indicator of the degree of familial clustering, and is a comparison of the risk of disease in the sibling of a patient compared to the risk for an individual in the general population. The λs value (11–53) in the Turkish population was reported to be higher than for a variety of other diseases considered to have complex genetic origins, including type 1 diabetes mellitus and rheumatoid arthritis. Although shared environmental factors could contribute to familial clustering, these high λs values unequivocally support a hereditary basis to Behçet’s disease. Further evidence for hereditary risk factors is the finding of genetic anticipation in Behçet’s disease, in which earlier disease onset is encountered in children of affected parents.39
HLA-B51 and the Silk Road
A heritable risk factor for Behçet’s disease was first identified in 1982, when an association with HLA-B5 in the human MHC was first reported.40 The HLA-B*5 locus includes the family of HLA-B51 alleles and HLA-B52. In the great majority of racial groups, the greatest heritable risk factor for disease, and indeed for ocular disease severity, is HLA B51.41 Recent data indicate that while at least 21 different alleles may exist at the HLA-B*51 locus (B*5101, 5102, etc—asterix denoting modern molecular typing), both B*5101 and B*5108 are associated with disease.42 However, HLA-B*52, which differs from HLA-B*51 by only two amino acids in the peptide binding groove, is not associated with Behçet’s disease in any population, suggesting that selective peptide binding, which is expected to differ between B*51 and B*52, may influence disease risk. However, it remains unclear which specific peptides, if any, are bound by B*51.
The geographical distribution of disease, encountered mainly in countries spanning the Mediterranean basin and the Far East, led to the synonym of Silk Road disease. These regions lie between latitudes 30° and 45° north, and it is of interest that the reported prevalence of Behçet’s disease is associated with latitude and is rarely encountered in more northern climes, the Americas, and Australasia29 (Table 3). Furthermore, the geographical distribution of HLA-B*51 among healthy subjects roughly corresponds with global disease distribution (Fig 5); in countries in the southern hemisphere (for example, Africa, South Pacific), and in Europe above 45° N, the prevalence of HLA-B*51 in healthy individuals is low or nil, with a similarly low reported rate of Behçet’s disease. This association between geographical disease distribution and HLA-B*51 has invoked the widely held belief that genetic risk factors were propagated by migrant traders along the Silk Road 2000 years ago. However, a more likely explanation is that the global distribution of associated gene(s) such as B*51 reflects not the passage of traders, but the earlier demographic movements across Asia and the Beringia Landmass (now the Beringia Straits) some 10 000 to 30 000 years ago. Since Africa, Eurasia, and the Americas had been populated by Homo sapiens by 300 000 bc, the virtual absence of HLA-B*51 in certain indigenous peoples (South Pacific and South American populations) indicates that HLA-B*51, and genes in linkage with B*51, must have been spread from West to East by subsequent migrations of peoples whose path broadly reflects the global distribution of Behçet’s disease.29
Figure 5.

Global distribution of HLA-B51 among healthy ethnic control groups with reference to the Silk Road and early demographic movements.
Thus, where HLA-B*51 is common, Behçet’s disease is encountered, and where this allele is rare, so too is disease. Indeed, a recent study of eight microsatellite markers in the MHC region in three dissimilar ethnic groups confirmed that the only significant association with disease for all groups was with HLA-B*51, leading to the conclusion that HLA-B*51 itself is the pathogenic gene.43 However, HLA-B*51 cannot be the sole causative factor for the simple reason that roughly a third of patients, even in countries with a high disease prevalence, do not possess this gene. Furthermore, the risk, or odds ratio (OR), of Behçet’s disease in an individual positive for HLA-B*51 shows a wide variation across Europe and Asia.29 A possible explanation for these data is that the HLA-B*51 molecule expresses the Bw4 motif, which itself may be causally related to disease, and which is found in other HLA types variably associated with Behçet’s disease, including HLA-B*15, HLA-B*27, and HLA-B*57.44,45 However, convincing evidence for a direct role for any of these molecules in Behçet’s disease is still awaited, and an alternative explanation is that these genetic domains are identified as markers for other associated “disease genes” within the MHC, as discussed below.
Other MHC associations with Behçet’s disease
Tumour necrosis factor
Over the past 20 years many new genes have been identified between HLA-B and the complement cluster. This area is densely packed with genes which encode proteins with immune related functions, including the complement system proteins, tumour necrosis factor (TNF), the heat shock protein (HSP) family, and the MHC class I chain related genes (MIC) (Fig 6). Certain allelic variants within these genes (polymorphisms) are in linkage disequilibrium with class I alleles (that is, these genes are more likely to be inherited than occur by chance alone), and their influence on the immune system has generated considerable interest in the context of MHC related inflammatory diseases.46 TNF is one such example of a gene domain close to the HLA class I region, and the contribution of TNF gene polymorphisms to MHC associated diseases has been thoroughly investigated. In 1992, an association between Behçet’s disease and alleles in the TNF promotor region was first reported in Japanese patients with Behçet’s disease,47 an association that has recently been confirmed among Middle Eastern patients.41 Two possible alleles may exist at the promotor site: TNFB*1 and TNFB*2. The latter is associated with higher TNF production by stimulated monocytes than the former and is not only more prevalent among patients with Behçet’s disease but is also weakly associated with a poor visual outcome.41 Although neither allele is rare, TNFB*2 has been associated with higher leucocyte TNF production, and might therefore lead to a more severe and prolonged inflammatory response. However, the TNFB*2 allele is also in linkage with HLA-B51, and these two alleles may therefore be co-inherited, each contributing both to disease risk and to severity of organ involvement.
Figure 6.

The human major histocompatibility complex (MHC) on the short arm of chromosome 6. TAP = transporter of antigenic peptides; 21-OH = steroid 21-hydroxylase enzyme; C4A and B = complement loci; Bf = properdin factor B of the alternate complement pathway; C2 = complement C2; TNF = tumour necrosis factor; MICA = MHC class I chain related gene; LT = lymphotoxin.
MHC class I chain related (MIC) gene
A second gene in linkage with HLA-B51 is the MHC class I chain related (MIC) gene. The family of MIC genes, first reported in 1994, is situated in the region between TNF and HLA-B genes.48 The MICA gene encodes a polypeptide that is expressed predominantly on gastrointestinal epithelial cells, and has a predicted amino acid sequence similar to that of MHC class I molecules. This may be relevant in Behçet’s disease, since oral and oesophageal lesions are a common and early feature, and MICA molecules are recognised by γδ T cells and natural killer (NK) cells. This is of interest because both γδ and NK cells have cytotoxic function and are increased in peripheral blood of patients with Behçet’s disease,49 and γδ cells have also been recovered from the aqueous humour of patients.50 Moreover, a significantly increased proportion of γδT cells express activation markers (CD69, CD29), produce IFNγ and TNF, and in particular are upregulated in response to peptides derived from Hsp 65 in patients compared to controls.51–53
An association between MICA genes and Behçet’s disease was first described in 1997, when it was shown that the prevalence of a certain “triplet repeat” polymorphism in the transmembrane region of the MICA gene (the MICA A6 allele) was raised in Japanese patients with Behçet’s disease.54 In 1999, these findings were confirmed in a Middle Eastern population, and a second MICA gene, encoding part of the extracellular region of the molecule (the MIC 009 allele), was also found to be more prevalent among patients.55 The initial inference was that certain MICA alleles might influence MICA function, leading to abnormal interactions between infected epithelial cells and cytotoxic T cells in Behçet’s disease. Hopes that MICA might be the long awaited Behçet’s disease “susceptibility gene” faded, however, when subsequent analysis revealed a tight linkage between these alleles and HLA-B51, which itself remained the strongest genetic risk factor for disease. Behçet’s disease cannot therefore be attributed to MICA alleles alone; nevertheless the position of MICA proteins in the GI tract and their recognition by γδ T cells strongly indicates that the inheritance of HLA-B51 linked MICA alleles may also influence disease risk (Fig 7). In support of a role in Behçet’s disease, γδT cells have been shown to proliferate in response to supernatant from cultured micro-organisms obtained from an oral ulcer from a patient with Behçet’s disease compared to healthy controls.56 In addition to the response to micro-organisms, γδT cells from patients also recognise the non-peptide prenyl pyrophosphate antigen,50 indicating that γδ T cells in Behçet’s disease are in a primed state and may be involved in mediating pathogenesis.
Figure 7.

Behçet’s disease. Putative role of MICA molecules and HSP expressed on mucosal epithelial cells. *The MHC class I chain related (MIC) gene locus is situated adjacent to the HLA-B domain. MICA is of interest in Behçet’s disease because it is expressed at gastrointestinal epithelial surfaces in response to bacterial infection. γδ T cells and NK cells are upregulated in Behçet’s disease and are known to recognise and kill MICA transfected cells. Recently, MICA was shown to be a ligand for NKG2D which is also expressed on γδ T cells and NK cells. †Heat shock proteins (HSP) are expressed in response to cellular shock, and have been found on cells lining the gastointestinal tract. T cell responses to HSP derivatives are reported to be raised in patients, and the genetic promoter region for HSP expression is also known to control MICA expression.
NEW INSIGHTS INTO THE GENETIC BASIS OF BEHçET’S DISEASE
In recent years, newly identified non-MHC regions have been reported as risk factors for Behçet’s disease. These discoveries further support a multigenic aetiology, and may disclose new therapeutic avenues.
Factor V Leiden and venous occlusion in Behçet’s disease
Behçet’s disease confers up to a 14-fold risk of developing systemic venous thrombosis (intracranial example, Fig 8), and the risk in males is reported to be six times higher than in females.57 In the eye, recurrent retinal vascular occlusion may occur in up to 48% of all ocular patients and is a major cause of visual loss in Behçet’s disease. In 1994, a point mutation in the factor V gene on chromosome 1 was discovered in Leiden, Holland (the Leiden mutation) and is now recognised as the most common hereditary abnormality of the clotting system.58 It is a risk factor for idiopathic systemic venous thrombosis and has also been associated with Behçet’s disease, in which systemic venous thrombosis occurs in up to 40% of all patients. Since 1996, this mutation has been reported to be raised among patients in Saudi Arabia59 and Turkey.60 More recently the mutation been associated with ocular disease, and in particular the development of ocular vaso-occlusion.61 This discovery indicates that genes outside the MHC may contribute independently to morbidity in Behçet’s disease, and raises the question of whether anticoagulation in ocular patients with aggressive disease may play a part in managing these patients.
Figure 8.

Intracranial venous thrombosis. (A) Male patient presents with headaches and disc oedema. (B) Magnetic resonance imaging and (C) magnetic resonance angiography demonstrating superior sagittal sinus thrombosis.
Intercellular adhesion molecule polymorphisms
The intercellular adhesion molecules (ICAM) are a family of receptors expressed on the surface of activated vascular endothelium. These molecules interact with their ligand molecules on leucocytes, enabling the movement of pro-inflammatory cells across the vessel wall into the tissues (see Fig 9). Once released from the endothelial surface, the soluble form of ICAM-1 (sICAM-1) may be detected in the peripheral blood of patients with a variety of inflammatory disorders, including Behçet’s disease.62 The ICAM-1 gene is encoded on chromosome 19 and contains at least two positions of allelic variation. These sites probably encode structural parts of the ICAM-1 molecule, and variations in the DNA sequences at these positions may therefore affect the binding properties between ICAM-1 and its leucocyte receptors. Alleles at both positions have been associated with inflammatory diseases such as multiple sclerosis,63 inflammatory bowel disease,64 and renal allograft failure,65 and, more recently with Behçet’s disease.66,67 However, until the effect of these allelic variants on the function of ICAM-1 is known, the relevance of these findings to Behçet’s disease remains unclear. Furthermore, since the ICAM-1 gene alleles in question are common in control populations, their relative effect on ICAM-1 binding and their influence on the risk of developing Behçet’s disease are likely to be small. Nevertheless, these findings also support a multigenic disease aetiology.
Figure 9.

Behçet’s disease. Factors affecting endothelial cell activation and leucocyte extravasation.
Behçet’s disease, familial Mediterranean fever, and genetic control of neutrophils
Familial Mediterranean fever (FMF) shares a number of features in common with Behçet’s disease. It occurs commonly in people of Arab, Turkish, or non-Ashkenasi Jewish origin, is characterised by recurring bouts of fever, peritonitis, arthritis, pleurisy and skin rashes, and may also lead to renal amyloidosis. Recently the gene associated with FMF was identified (MEFV), and the disease has been shown to be inherited in an autosomal recessive manner.68,69 The gene codes for a protein called pyrin, which is thought to be involved in regulating neutrophils in the inflammatory response. Several mutations in the MEFV gene have been identified and, given the ethnic distribution of FMF and the involvement of neutrophils in both diseases, these have been studied in patients with Behçet’s disease. Four mutations have been shown to be more prevalent in patients with Behçet’s disease (range 2.6%–10.5%) compared with control individuals (range 0%–2.2%) suggesting that MEFV mutations may act as susceptibility factors in Behçet’s disease.70 Moreover, MEFV mutations present in the Middle East have never been reported in Japan, and it has been suggested that this might explain why amyloidosis seen in Behçet’s disease follows the same geographical pattern.71 Finally, it is of interest that in those unfortunate individuals with a diagnosis of both FMF and Behçet’s disease, only one MEFV gene mutation has been identified, suggesting that Behçet’s disease can precipitate FMF (usually a recessive disease), in individuals with only a single MEFV mutation.72 Collectively, these findings indicate that (a)Behçet’s disease activation of PMN is sufficient to overcome the heterozygous state of FMF carriers, and (b) an abnormal genetic influence on neutrophils which is responsible for FMF may also play a part in Behçet’s disease.
Killing inhibitory receptors (KIR)
The most fundamental question in Behçet’s disease research is how HLA-B51 is causally related to the inflammatory events at the mucosal and/or vascular endothelial surfaces. Recently, a new family of T cell receptors, expressed mainly by natural killer (NK) cells and γδ T cell receptor positive T cells (both implicated in Behçet’s disease) has been described.73 This family of receptors is termed killer inhibitory receptors (KIR), and engagement of KIRs is known to inhibit cell mediated toxicity.74 One KIR (NKB1) binds to molecular sequences which are also found in some HLA class I molecules, including HLA-B51. An expansion of CD8+ cells, γδ TCR+ cells, and NK cells has been reported in patients with Behçet’s disease, and more recently defective expression of KIR on NK cells in patients with Behçet’s disease has also been reported.75 This has led to the suggestion that the host MHC class I haplotype (for example, HLA-B*51) may “fine tune” the regulation of KIR expression.76 Thus, KIR binding to sequences present in HLA-B51 expressed on gastrointestinal epithelial cells in patients with Behçet’s disease may lead to a defect in the regulation of T cell inhibition or activation, and could in turn lead to abnormal responses to microbiological challenges at the epithelial surface.
SUMMARY
Huluci Behçet himself postulated a viral trigger for this disease. Sixty five years later it remains unclear whether a virus or bacterium initiates and/or prolongs the characteristic mucosal and endothelial hyper-reactivity observed at the gastrointestinal and vascular endothelial surfaces, respectively. However, it has become increasingly apparent that these events, once triggered, may be influenced by numerous interdependent and independent genetic regions. Despite 20 years of intense efforts to identify other associated genetic regions in chromosome 6 and elsewhere, HLA-B51 remains foremost among candidate risk factors for disease (Fig 10). TNF and MICA alleles in the MHC are also implicated, but their close linkage with HLA-B51 has made their independent contribution to disease less easy to define. Newly characterised regions on different chromosomes may also contribute to disease, through their influence on the clotting cascade, on the events that underlie the leucocyte recruitment into the tissues, and on neutrophil activation. These discoveries have important implications for the management of patients with Behçet’s disease. Firstly, they provide a rationale for new forms of immunomodulation that are now under investigation such as TNF and ICAM blockade, and interferon α-2a. In the case of factor V Leiden, anticoagulative measures, in addition to immunosuppression, may be valuable in managing patients with severe and recurrent ocular occlusive disease. Secondly, increasing knowledge of genetic disease severity markers may enable early identification of vulnerable patients before irreversible ocular complications ensue. This, in association with increasingly effective and targeted immune blockade, has every chance of improving the outlook of this blinding disease.
Figure 10.

Behçet’s disease is associated with multiple hereditory and environmental risk factors.
Acknowledgments
This work was funded by the Iris Fund for the Prevention of Blindness, the TFC Frost Charitable Trust, and the St John Eye Hospital in Jerusalem.
REFERENCES
- 1.Behçet H. Über rezidiverende, apthöse durch ein Virus verursachte Geschwüre am Mund, am Auge und an der Genitalen. Dermatol Wochenschr 1937;105:1152–7. [Google Scholar]
- 2.Adamantiades B. Sur un case d’iritis á hypopyon récidivant. Ann Oculist (Paris) 1931;168:271–8. [Google Scholar]
- 3.Feigenbaum A. Description of Behçet’s Syndrome in the Hippocratic Third Book of Endemic Diseases. Br J Ophthalmol 1956;40:355–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Study Group for Behçet’s Disease. Criteria for diagnosis of Behçet’s disease. Lancet 1990;335:1078–80. [PubMed] [Google Scholar]
- 5.Lee E-S, Hur W, Bang D, et al. Prognosis of recurrent oral ulceration in Behçet’s disease. In: Godeau P, Wechsler B, eds. Behçet’s disease. Paris: Elsevier, 1993:291–4.
- 6.Dilsen N, Konice M, Aral O, et al. Risk factors for vital organ involvement in Behçet’s disease. In: Godeau P, Wechsler B, eds. Behçet’s disease. Paris: Elsevier, 1993:165–9.
- 7.Lueck CJ, Pires M, McCartney AC, et al. Ocular and neurological Behçet’s disease without orogenital ulceration? J Neurol Neurosurg Psychiatry 1993;56:505–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imai Y. Studies on prognosis and symptoms of Behçet’s disease in long-term observation. Jpn J Clin Ophthalmol 1971;25:661–94. [Google Scholar]
- 9.Chams C, Mansoori P, Shahram F, et al. Iconography of mucocutaneous lesions of Behçet’s disease. In: Godeau P, Wechsler B, eds. Behçet’s disease. Paris: Elsevier, 1993:359–62.
- 10.Matsuo T, Itami M, Nakagawa H, et al. The incidence and pathology of conjunctival ulceration in Behçet’s syndrome. Br J Ophthalmol 2002;86:140–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Howe L, Denman A, Dinning W, et al. Visual prognosis in Behçet’s disease. In: Godeau P, Wechsler B, eds. Behçet’s disease. Elselvier Science Publishers, 1993:599–602.
- 12.Muhaya M, Lightman S, Ikeda E, et al. Behçet’s disease in Japan and Great Britain. Ocular Immunol Inflamm 2000;8:141–8. [PubMed] [Google Scholar]
- 13.Whitcup SM, Salvo EC Jr, Nussenblatt RB. Combined cyclosporine and corticosteroid therapy for sight-threatening uveitis in Behcet’s disease. Am J Ophthalmol 1994;118:39–45. [DOI] [PubMed] [Google Scholar]
- 14.Kilmartin DJ, Forrester JV, Dick AD. Tacrolimus (FK506) in failed cyclosporin A therapy in endogenous posterior uveitis. Ocul Immunol Inflamm 1998;6:101–9. [DOI] [PubMed] [Google Scholar]
- 15.Sloper CM, Powell RJ, Dua HS. Tacrolimus (FK506) in the treatment of posterior uveitis refractory to cyclosporine. Ophthalmology 1999;106:723–8. [DOI] [PubMed] [Google Scholar]
- 16.Hamuryudan V, Ozyazgan Y, Hizli N, et al. Azathioprine in Behçet’s syndrome: effects on long-term prognosis. Arthritis Rheum 1997;40:769–74. [DOI] [PubMed] [Google Scholar]
- 17.Goossens PH, Verburg RJ, Breedveld FC. Remission of Behcet’s syndrome with tumour necrosis factor alpha blocking therapy. Ann Rheum Dis 2001;60:637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sfikakis PP, Theodossiadis PG, Katsiari CG, et al. Effect of infliximab on sight-threatening panuveitis in Behçet’s disease. Lancet 2001;358:295–6. [DOI] [PubMed] [Google Scholar]
- 19.Triolo G, Vadala M, Accardo-Palumbro A, et al. Anti-tumour necrosis factor monoclonal antibody treatment for ocular Behçet’s disease. Ann Rheum Dis 2002;61:560–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stuebiger N, Koetter I, Zierhut M. Complete regression of retinal neovascularization after therapy with interferon alpha in Behcet’s disease. Br J Ophthalmol 2000;84:1437–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kotter I, Zierhut M, Eckstein AK, et al. Human recombinant interferon alfa-2a for the treatment of Behcet’s disease with sight threatening posterior or panuveitis. Br J Ophthalmol 2003;87:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Metzger R, Heckl-Ostreicher B, Nerl C, et al. Immunological studies of gamma delta T cells in a case of large granular lymphocyte (LGL) leukemia: leukemic gamma delta+ T cells are resistant to growth stimulation in vitro but respond to interferon-alpha treatment in vivo. Leuk Res 1992;16:1087–95. [DOI] [PubMed] [Google Scholar]
- 23.Karti SS, Ovali E, Ratip S, et al. Effect of interferon-alpha (2a) on neutrophil adhesion and phagocytosis in chronic myeloid leukeamia and Behcet’s disease. Clin Rheumatol 200:21;211–14. [DOI] [PubMed] [Google Scholar]
- 24.Ergun T, Gurbuz O, Harvell J, et al. The histopathology of pathergy: a chronologic study of skin hyperreactivity in Behçet’s disease. Int J Dermatol 1998;37:929–33. [DOI] [PubMed] [Google Scholar]
- 25.Demirkesen C, Tuzuner N, Mat C, et al. Clinicopathologic evaluation of nodular cutaneous lesions of Behçet’s syndrome. Am J Clin Pathol 2001;116:341–6. [DOI] [PubMed] [Google Scholar]
- 26.Tugal-Tutkun I, Urgancioglu M, Foster CS. Immunopathologic study of the conjunctiva in patients with Behçet’s disease. Ophthalmology 1995;102:1660–8. [DOI] [PubMed] [Google Scholar]
- 27.Mege J-L, Dilsen N, Sanguedolce V, et al. Overproduction of monocyte derived TNF, Interleukin (IL) 6, IL 8 and increased neutrophil superoxide generation in Behçet’s disease. A comparative study with familial Mediterranean fever and healthy subjects. J Rheumatol 1993;20:1544–9. [PubMed] [Google Scholar]
- 28.Accardo-Palumbro A, Triolo G, Carbone MC, et al. Polymorphonuclear leukocyte myeloperoxidase levels in Behçet’s disease. Clin Exp Rheumatol 2000;18:495–8. [PubMed] [Google Scholar]
- 29.Verity DH, Vaughan RW, Marr JE, et al. Behçet’s disease, The Silk Road and HLA-B51: historical and geographical perspectives. Tissue Antigens 1999;54:213–20. [DOI] [PubMed] [Google Scholar]
- 30.Parker G, ed. The commercial and cultural bonds of Eurasia. In: The Times atlas of world history. 4th ed. London: Times Books, 1993.
- 31.Kaneko F, Oyama N, Nishibu A. Streptococcal infection in the pathogenesis of Behçet’s disease and clinical effects of minocycline on the disease symptoms.Yonsei Med J 1997;38:444–454. [DOI] [PubMed] [Google Scholar]
- 32.Lehner T. The role of heat shock protein, microbial and autoimmune agents in the aetiology of Behçet’s disease. Int Rev Immunol 1997;14:21–32. [DOI] [PubMed] [Google Scholar]
- 33.Direskeneli H, Eksioglu-Demiralp E, Yavuz S, et al. T cell responses to 60/65 kDa heat shock protein derived peptides in Turkish patients with Behcet’s disease. J Rheumatol 2000;27:708–13. [PubMed] [Google Scholar]
- 34.Bang D, Yoon KH, Chung HG, et al. Epidemiolgical and clinical features of Behçet’s disease in Korea. Yonsei Med J 1997;38:428–36. [DOI] [PubMed] [Google Scholar]
- 35.Calguneri M, Ertenli I, Kiraz S, et al. Effect of prophylactic benzathine penicillin on mucocutaneous symptoms of Behçet’s disease. Dermatology 1996;192:125–8. [DOI] [PubMed] [Google Scholar]
- 36.Lehner T. The role of heat shock protein, microbial and autoimmune agents in the aetiology of Behçet’s disease. Int Rev Immunol 1997;14:21–32. [DOI] [PubMed] [Google Scholar]
- 37.Sohn S, Lee ES, Bang D, et al. Behcet’s disease-like symptoms induced by the Herpes simplex virus in ICR mice. Eur J Dermatol 1998;8:21–3. [PubMed] [Google Scholar]
- 38.Gul A, Inanc M, Ocal L, et al. Familial aggregation of Behcet’s disease in Turkey. Ann Rheum Dis 2000;59:622–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fresko I, Soy M, Hamurydan V, et al. Genetic anticipation in Behçet’s syndrome. Ann Rheum Dis 1998;57:45–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ohno S, Ohguchi M, Hirose S, et al. Close association of HLA-Bw51 with Behçet’s disease. Arch Ophthalmol 1982;100:1455–8. [DOI] [PubMed] [Google Scholar]
- 41.Verity DH, Wallace GR, Vaughan RW, et al. HLA and TNF polymorphisms in ocular Behçet’s disease. Tissue Antigens 1999;54:264–72. [DOI] [PubMed] [Google Scholar]
- 42.Mizuki N, Ota M, Katsuyama Y, et al. HLA class I genotyping including HLA-B*51 allele typing in the Iranian patients with Behçet’s disease. Tissue Antigens 2001;57:457–62 [DOI] [PubMed] [Google Scholar]
- 43.Mizuki N. Ota M. Yabuki K, et al. Localization of the pathogenic gene of Behçet’s disease by microsatellite analysis of three different populations. Invest Ophthmol Vis Sci 2000;41:3702–8. [PubMed] [Google Scholar]
- 44.Choukri F, Chakib A, Himmich H, et al HLA-B*51 and B*15 alleles confer predisposition to Behcet’s disease in Moroccan patients. Hum Immunol 2001;62:180–5. [DOI] [PubMed] [Google Scholar]
- 45.Gul A, Uyar FA, Inanc M, et al. A weak association of HLA-B*2702 with Behçet’s disease. Genes Immun. 2002;3:368. [DOI] [PubMed] [Google Scholar]
- 46.Ahmad T, Wallace GR, James T, et al. Mapping the HLA association in Behçet’s disease: a role for tumour necrosis factor polymorphisms. Arthritis Rheum 2003;48:807–13. [DOI] [PubMed] [Google Scholar]
- 47.Mizuki N, Inoko H, Sugimura K, et al. RFLP analysis in the TNF β gene and susceptability to alloreactive NK cells in Behçet’s disease. Inv Ophthalmol Vis Sci 1992:33:3084–90. [PubMed] [Google Scholar]
- 48.Bahram S, Bresnahan M, Geraghty DE, et al. A second lineage of mammalian major histocompatibility complex class I genes. Proc Natl Acad Sci USA 1994;91:6259–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nishida T, Hiruyama K, Nakamura S, et al. Proliferative response of CD8+ γδ T cells to Streptococcus sanguis in patients with Behçet’s disease. Ocular Immunol Inflam 1998;6:139–44 [DOI] [PubMed] [Google Scholar]
- 50.Verjans GM, van Hagen PM, van der Kooi A, et al. Vgamma9Vdelta T cells recovered from eyes of patients with Behçet’s disease recognize prenyl pyrophosphate antigens. J Neuroimmunol 2002;130:46–54. [DOI] [PubMed] [Google Scholar]
- 51.Eksioglu-Demiralp E, Direskeneli H, Ergun T, et al. Increased CD4+CD16+ and CD4+CD56+ T cell subsets in Behçet’s disease. Rheumatol Int 1999;19:23–6. [DOI] [PubMed] [Google Scholar]
- 52.Freysdottir J, Lau SH, Fortune F. Gamma-delta+ T cells in Behçet’s disease (BD) and recurrent aphthous stomatitis (RAS). Clin Exp Immunol 1999;118:451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hasan A, Fortune F, Wilson A, et al. Role of γδ T cells in pathogenesis and diagnosis of Behçet’s disease. Lancet 1996;347:789–93. [DOI] [PubMed] [Google Scholar]
- 54.Mizuki N, Ota M, Kimura S, et al. Triplet repeat polymorphism in the transmembrane region of the MICA gene: a strong association of six GCT repetitions with Behçet’s disease. Proc Natl Acad Sci USA 1997;94:1298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wallace GR, Verity DH, Delamaine L, et al. Association with MICA alleles with Behçet’s disease. Immunogenetics 1999;49:613–17. [DOI] [PubMed] [Google Scholar]
- 56.Bank I, Duvdevani M, Livneh A. Expansion of gamma-delta T cells in Behçet’s disease: role of disease activity and microbial flora in oral ulcers. J Lab Clin Med 2003;141:33–40. [DOI] [PubMed] [Google Scholar]
- 57.Ames PR, Steuer A, Pap A, et al. Thrombosis in Behçet’s disease: a retrospective survey from a single UK centre. Rheumatology 2001;40:652–5. [DOI] [PubMed] [Google Scholar]
- 58.Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64–7. [DOI] [PubMed] [Google Scholar]
- 59.Mammo L, Al-Dalaan A, Bahabri SS, et al. Association of factor V Leiden with Behçet’s disease. J Rheumatol 1997;24:2196–8. [PubMed] [Google Scholar]
- 60.Gül A, Ozbeck U, Ozturk C, et al. Coagulation factor V gene mutation increases the risk of venous thrombosis in Behçet’s disease. Br J Rheumatol 1996;35:1178–80. [DOI] [PubMed] [Google Scholar]
- 61.Verity DH, Vaughan RW, Madanat W, et al. Factor V Leiden mutation is associated with ocular involvement in Behçet’s disease. Am J Ophthalmol 1999;128:352–6. [DOI] [PubMed] [Google Scholar]
- 62.Verity DH, Wallace GR, Seed P, et al. Soluble adhesion molecules in Behçet’s disease. Ocul Immunol Inflamm 1998;6:81–92. [DOI] [PubMed] [Google Scholar]
- 63.Mycko MP, Kwinkowski M, Troncznska E, et al. Multiple sclerosis: the increased frequency of the ICAM-1 exon 6 gene point mutation genetic type K469. Ann Neurol 1998;44:70–5. [DOI] [PubMed] [Google Scholar]
- 64.Yang H, Vora DK, Targan SR, et al. Intercellular adhesion molecule 1 gene associations with immunologic subsets of inflammatory bowel disease. Gastroenterology 1995;109:440–8. [DOI] [PubMed] [Google Scholar]
- 65.McLaren AJ, Marshall SE, Haldar NA, et al. Adhesion molecule polymorphisms in chronic renal allograft failure. Kid Int 1999;55:1977–82. [DOI] [PubMed] [Google Scholar]
- 66.Verity DH, Vaughan RW, Kondeatis E, et al. Intercellular adhesion molecule-1 gene polymorphisms in Behçet’s disease. Eur J Immunogen 2000;27:73–6. [DOI] [PubMed] [Google Scholar]
- 67.Boiardi L, Salvarani C, Casali B, et al. Intercellular adhesion molecule-1 gene polymorphisms in Behçet’s disease. J Rheumatol 2001;28;1283–7. [PubMed] [Google Scholar]
- 68.The French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997;17:25–31. [DOI] [PubMed] [Google Scholar]
- 69.The International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997;90:797–807. [DOI] [PubMed] [Google Scholar]
- 70.Touitou I, Magne X, Molinari N, et al. MEFV mutations in Behçet’s disease. Hum Mutation 2000;16:271–2. [DOI] [PubMed] [Google Scholar]
- 71.Akpolat T, Yilmaz E, Akpolat I, et al. Amyloidosis in Behçet’s disease and familial Mediterranean fever. Rheumatology 2002;41:592–3. [DOI] [PubMed] [Google Scholar]
- 72.Livneh A, Aksentijevich I, Langevitz P, et al. A single mutated MEFV allele in Israeli patients suffering from familial Mediterranean fever and Behçet’s disease (FMF-BD). Eur J Hum Genet 2001;9:191–6. [DOI] [PubMed] [Google Scholar]
- 73.Lanier LL. NK cell receptors for polymorphic MHC class I molecules. In: Charron D, ed. Genetic diversity of HLA. Functional and medical implications. Proceedings of the Twelth International Histocompatibility Workshop and Conference. Volumes I and II. Sevres, France: EDK Publishing, 1997.
- 74.Steinle A, Li P, Morris DL, et al. Interactions of human NKG2D with its ligands MICA, MICB, and homologs of the mouse RAE-1 protein family. Immunogenetics 2001:53;279–87. [DOI] [PubMed] [Google Scholar]
- 75.Shimoyama Y, Takeno M, Kashiwakura J, et al. Reduced KIR expression is associated with NKT cell expansion in patients with Behçet’s disease (abstract 1318). Arthritis Rheum 1998;41:S252. [Google Scholar]
- 76.Mingari MC, Moretta A, Moretta L. Regulation of KIR expression in human T cells: a safety mechanism that may impair protective T cell responses. Immunol Today 1998;19:153–7. [DOI] [PubMed] [Google Scholar]