

Abstract
Aim: To describe the phenotype of a three generation consanguineous Pakistani family containing six individuals with autosomal recessive cone dystrophy caused by mutation in GNAT2.
Methods: Five of the six affected individuals underwent an ophthalmological examination, electrodiagnostic testing, fundus photography, autofluorescence imaging, and detailed psychophysical testing.
Results: All five examined patients had a history of nystagmus from infancy, photophobia, defective colour vision, and poor visual acuity. The nystagmus in three of the individuals had lessened with time. Fundus examination revealed an abnormal foveal appearance, without frank atrophy or pigmentation. Electroretinography (ERG) revealed absent ISCEV cone flicker ERGs with some preservation of responses to short wavelength stimulation. Rod ERGs showed no definite abnormality, but maximal (mixed rod-cone) response a-wave amplitudes were mildly subnormal. Rudimentary residual colour vision was detected in three individuals. There is clinical evidence of progressive visual acuity reduction in two older individuals.
Conclusion: Mutation in the α-subunit of cone specific transducin (GNAT2) is characterised by an infantile onset cone dystrophy. Some affected individuals may show deterioration of visual acuity with time.
Keywords: cone dystrophy, phenotype, transducin
The cone and cone-rod dystrophies form part of a clinically heterogeneous group of retinal dystrophies that are a major cause of childhood blindness. The major clinical features of cone dystrophies are reduced visual acuity, abnormal colour vision, photophobia, central scotomata, and often nystagmus. Cone dystrophies have been described with autosomal dominant, autosomal recessive, or X linked patterns of inheritance.1,2
Cone and cone-rod dystrophies are also phenotypically heterogeneous.2 Various subtypes have been identified on the basis of natural history and psychophysical and electrophysiological testing.3–5 These disorders may be stationary or progressive. The two well characterised stationary cone dystrophies are blue cone monochromatism, an X linked disorder in which there are only two functional classes of photoreceptor (rods and S-cones), and rod monochromatism.
Rod monochromatism or complete achromatopsia is a stationary cone dystrophy, with an incidence of approximately 1 in 30 000, in which there is an absence of functioning cone photoreceptors.6,7 Affected individuals usually present in infancy with nystagmus, poor visual acuity, photophobia, and complete colour blindness. Fundus examination is usually normal, but electroretinography reveals absent photopic (cone) responses and normal scotopic (rod) responses. Individuals with incomplete achromatopsia retain some colour vision.
Achromatopsia is recessively inherited and genetically heterogeneous. To date, three achromatopsia genes have been characterised, the first two described being CNGA38–10 and CNGB3,11–13 located at 2q11 and 8q21 respectively. CNGA3 and CNGB3 code for the α and β subunits of the cGMP gated cation channel in cone cells, respectively. The gene coding for the α-subunit of cone specific transducin (GNAT2) was proposed as a candidate gene for achromatopsia by Mollon in 1997,14 and mutations in this gene have recently been described in patients with achromatopsia.15,16 However, a detailed description of the phenotype associated with GNAT2 inactivation has not been presented. In this report we have reviewed the phenotype of the large consanguineous Pakistani family in whom we identified a novel frameshift mutation in GNAT2 (c842_843insTCAG; M280fsX291).16
PATIENTS AND METHODS
Five affected members of a three generation, consanguineous Pakistani family with cone dystrophy were assessed after informed consent was obtained (Fig 1).
Figure 1.

Family pedigree. Solid symbols indicate clinically affected individuals and open symbols represent unaffected individuals.
A full medical history was taken and an ophthalmological examination performed. Examined subjects also underwent colour fundus photography and fundus autofluorescence imaging using the confocal scanning laser ophthalmoscope (cSLO) (Zeiss Prototype; Carl Zeiss Inc, Oberkochen, Germany). Electrodiagnostic assessment included electro-oculography (EOG), full field electroretinography (ERG) and pattern ERG (PERG), incorporating the protocols recommended by the International Society for Clinical Electrophysiology of Vision.17–19 S-cone ERGs were also recorded using a previously described protocol.20
Colour vision testing included the use of the Hardy, Rand, Rittler (HRR) plates (American Optical Company, New York, NY, USA), Sloan achromatopsia plates, enlarged Farnsworth D-15 (PV-16), the enlarged Mollon-Reffin (M-R) minimal test,21 and the Nagel anomaloscope. The PV-16, Sloan achromatopsia plates, and the M-R test were all performed under CIE Standard Illuminant C from a MacBeth Easel lamp.
The PV-16 and the enlarged M-R test were used in order to detect any residual colour discrimination that might be present in patients with low vision: the coloured discs of the PV-16 were 33 mm in diameter and those of the enlarged M-R test were 26 mm in diameter (corresponding to visual angles of 3.8 and 3.3 deg at a viewing distance of 500 mm). With the same purpose in mind, we also used a modification of the Cambridge colour test, a computerised test that allows the measurement of colour discrimination along different directions in colour space.22,23 In the modified test, the stimulus array consisted of only four large discs, organised in a diamond pattern. Each disc subtended 4 degrees of visual angle at the viewing distance of 1 metre. On any presentation, one of the discs differed in chromaticity from the remaining three, and the patient’s task was to identify this disc by pressing one of four buttons within 4 seconds. To ensure that the discrimination was on the basis of chromaticity, the luminance of each disc was given a random value chosen from six levels between 4 cd/m2 and 24 cd/m2. To establish the patient’s threshold for a given direction in colour space, the chromatic difference between the target and distractor discs was adjusted by a double staircase procedure.24
RESULTS
All five patients had a history of nystagmus from infancy, mild photophobia, defective colour vision, and poor visual acuity (6/60 to CF). They all described improved vision in mesopic conditions. Examination of the anterior segment was unremarkable, except for one individual (VI:3) who had a unilateral congenital cataract. Fundus examination revealed a mildly abnormal foveal appearance but without frank atrophy or pigmentation (Fig 2). Peripheral retinal examination was normal in all subjects. ERG showed absent cone responses to 30 Hz flicker, small responses to short wavelength stimulation, and normal rod specific ERGs, but mildly subnormal maximal response a-wave amplitudes (Fig 3). Autofluorescence imaging was normal in all individuals. Clinical findings are summarised in Table 1.
Figure 2.
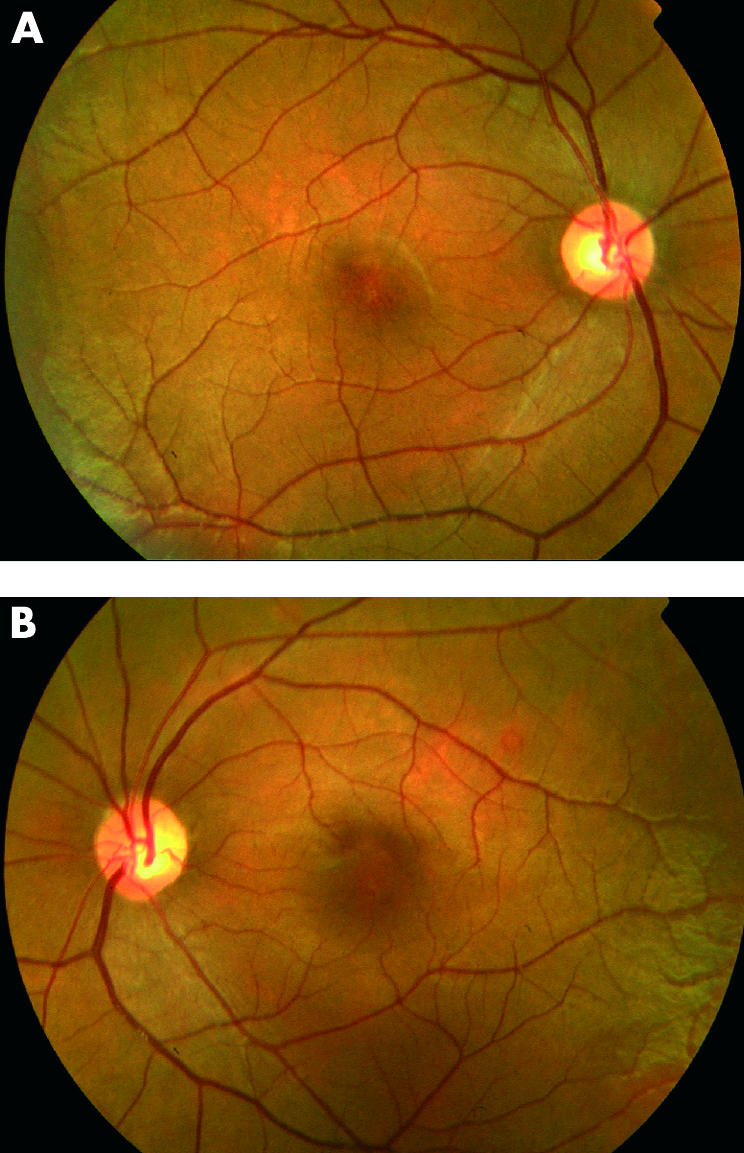
Fundus photographs. Mildly abnormal foveal appearance, but without frank atrophy or pigmentation.
Figure 3.

Electrophysiological data from two patients, a father (V:7) and son (VI:1). A normal control is shown for comparison. Note the differences in calibration between the normal and the two patients for the cone derived ERGs. Both patients have normal rod specific ERGs, with borderline subnormal a-wave in the maximal response of the son, mildly subnormal in the father. Flicker ERG is undetectable in both patients, but there is some very low amplitude activity with photopic single flash stimulation. S-cone specific stimulation, using a blue light superimposed on an orange background,20 suggests some preservation of mechanisms sensitive to short wavelengths. Note the presence of an earlier peak at ∼30 ms in the normal, absent in the two patients, which reflects activity from L/M cone systems.
Table 1.
Summary of clinical findings
| Patient | Age | VA | Refraction | Horizontal pendular nystagmus | ERG | Fundus | M-R colour vision test |
| V:2 | 35 | R CF | −3.5/+3.0×30 | Prominent | Absent 30 Hz cone responses; | Abnormal foveal appearance | P(no)D(no)T(no) |
| L CF | −4.0/+3.0×160 | Normal rod specific responses | P(no)D(no)T(no) | ||||
| V:4 | 41 | R 6/60 | −2.0/+2.0×110 | Absent | Absent 30 Hz cone responses; | Abnormal foveal appearance | P(no)D(no)T(no) |
| L 6/60 | −2.5/+1.5×70 | Normal rod specific responses | P(no)D(no)T(no) | ||||
| V:7 | 44 | R CF | −2.0/+2.0×135 | Prominent | Absent 30 Hz cone responses | Abnormal foveal appearance | P(no)D(7)T(5) |
| L CF | −1.5/+1.5 ×45 | Normal rod specific responses | P(no)D(6)T(5) | ||||
| VI:1 | 20 | R 6/60 | +1.5/+1.5×110 | Absent | Absent 30 Hz cone responses | Abnormal foveal appearance | P(no)D(7)T(4) |
| L 6/60 | +1.0/+2.0×80 | Normal rod specific responses | P(no)D(no)T(5) | ||||
| VI:3 | 19 | R CF | Balance | Minimal | Absent 30 Hz cone responses | Abnormal foveal appearance | NA |
| L 6/60 | −2.0/+3.0×75 | Normal rod specific responses | P(no)D(6)T(5) |
ERG = counting fingers; M-R = Mollon-Reffin test. The letters give the axis P = protan, D = deutan, and T = tritan. The number in parentheses gives the least saturated chip that could be discriminated from the greys; VI:3 has a right sided congenital cataract.
Two older individuals (V:2 and V:7) described a gradual deterioration of vision. Visual acuity in V:7 was 6/36 in both eyes when he first presented 30 years ago, while his current best corrected acuity is counting fingers. His brother V:2, also showed evidence of deterioration of visual acuity from 6/60 in both eyes documented 10 years ago, to counting fingers at present. Both subjects have more prominent horizontal pendular nystagmus than other affected family members.
All five patients had abnormal colour vision. At the Nagel anomaloscope all five patients exhibited a scotopic spectral sensitivity. Four patients displayed a scotopic pattern of arrangement on PV-16. One patient, V:7, unlike a typical patient with complete achromatopsia, produced only one major transposition on the PV-16. Rudimentary colour discrimination was also detected in three individuals on testing with the M-R minimal test, with variable ability to identify the most saturated chip along the deutan and/or tritan axes. Further evidence of residual colour vision was provided by the colour discrimination ellipses produced on computerised colour vision testing. The other two individuals, V:2 and V:4, displayed no residual colour vision and showed typical achromatopic matches on Sloan plates.
DISCUSSION
The phenotype in this family with a novel homozygous frameshift mutation in the cone α-transducin gene, GNAT2, is characterised by mild photophobia, nystagmus, abnormal colour vision, and poor visual acuity (6/36 to CF). Electroretinography using the ISCEV protocol revealed absent cone responses, with normal rod specific ERGs, but mild reduction in maximal response a-wave amplitudes. Small photopic responses to short wavelength stimulation were detectable. On detailed colour vision testing, residual colour discrimination was detected in three individuals.
This phenotype is similar therefore to the incomplete form of achromatopsia arising from certain mutations in CNGA3, the gene encoding the α-subunit of the cGMP gated cation channel in cones.10 Unlike complete achromatopsia, we have been able to record S-cone ERG responses in our patients and, in two older subjects, a worsening of visual acuity with age has been documented, although we have no definite evidence of progressive deterioration in retinal function. In achromatopsia we have found only one case report of progressive retinal degeneration in the form of mid-peripheral retinal pigmentation and concentric constriction of the peripheral visual fields.25 It was also reported that a few of the younger subjects in that achromatopsia series had small residual cone responses on ERG. Taken together, therefore, these findings may represent evidence that progression in retinal dysfunction may be present in at least some individuals with achromatopsia, but no natural history studies are available to corroborate this.25
Retinal dysfunction in our family is predominantly confined to cone photoreceptors. In cone cells, light activated photopigment interacts with transducin, a three subunit guanine nucleotide binding protein, stimulating the exchange of bound GDP for GTP. The cone α-transducin subunit (encoded by GNAT2), which is bound to GTP, is then released from its β and γ subunits and activates cGMP phosphodiesterase by removing the inhibitory γ subunits from the active site of this enzyme. cGMP phosphodiesterase lowers the concentration of cGMP in the photoreceptor which results in closure of cGMP gated cation channels and consequent hyperpolarisation of the photoreceptor.26 Thus, the finding of a germline GNAT2 mutation in a family with cone dystrophy is consistent with the known function of the GNAT2 product. Furthermore, mutations in human rod specific α-transducin, which is 83% homologous to cone α-transducin,27,28 have been shown to be associated with the Nougaret form of congenital stationary night blindness.29
The frameshift mutation identified in our family results in a truncated protein that lacks 63 amino acids from the carboxy terminal.16 All the GNAT2 mutations identified by Kohl et al would also result in premature translation termination and in protein truncation at the carboxy terminal.15 This region contains important functional domains of α-transducin which have been shown to interact with the rhodopsin30 and phosphodiesterase γ-subunits.31 However, if this mutation were to lead to a complete lack of α-transducin function, it is difficult to explain the residual colour vision along deutan and tritan axes in these individuals, if these are entirely cone mediated mechanisms. It is possible that the mutation results in a protein that, although severely reduced in efficacy, may still show some residual α-transducin function. Alternatively, there may be some redundancy within the cone phototransduction pathway that allows a level of continued function despite suboptimal or absent function of one of the components of the cascade.
The human cone transducin α-subunit (GNAT2) gene was completely characterised by Morris and Fong in 199327 and the evidence that this gene is expressed in all three cone types comes from the immunohistochemical demonstration that an antibody raised against cone α-transducin peptides crossreacts with all three classes of cone photoreceptor in the human retina.32 This does not however definitively rule out the possibility that S-cones may express an alternative form of α-transducin since identical epitopes may be present on both forms. It may also be significant that Southern blot analysis of human genomic DNA indicated that there may be more than one cone α-transducin gene.32 Therefore, although there are no subsequent studies that provide any direct evidence for an S-cone specific cone α-transducin, it remains a possibility that GNAT2 is not expressed in S-cones, and that the residual S-cone function detected in our family arises from the use of this other distinct form of α-transducin. In this case therefore, the residual tritan colour discrimination may be accounted for by a comparison of quantum catches in the remaining functional S-cones and rod photoreceptors, in the manner proposed to underlie colour discrimination detected in blue cone monochromatism.33
A detailed description of the phenotype associated with mutation in the α-subunit of cone specific transducin has not been previously reported. It is characterised by a cone dystrophy with an infantile onset, a deterioration of visual acuity with time in older individuals, and residual S-cone function.
Acknowledgments
The work was supported by grants from the British Retinitis Pigmentosa Society, the Guide Dogs for the Blind Association, and the Wellcome Trust. We are also grateful to the patients who kindly agreed to take part in this study.
REFERENCES
- 1.Goodman G, Ripps H, Siegel IM. Cone dysfunction syndromes. Arch Ophthalmol 1963;70:214–31. [DOI] [PubMed] [Google Scholar]
- 2.Simunovic MP, Moore AT. The cone dystrophies. Eye 1998;12:553–65. [DOI] [PubMed] [Google Scholar]
- 3.Pokorny J, Smith VC, Verriest G, et al. Congenital and acquired colour vision defects. New York: Grune and Stratton, 1979.
- 4.Szlyk J, Fishman GA, Alexander KR, et al. Clinical subtypes of cone-rod dystrophy. Arch Ophthalmol 1993;111:781–8. [DOI] [PubMed] [Google Scholar]
- 5.Sadowski B, Zrenner E. Cone and rod function in cone degenerations. Vis Res 1997;37:2303–2314. [DOI] [PubMed] [Google Scholar]
- 6.Sharpe LT, Nordby K. Total colour-blindness: an introduction. In: Hess RF, Sharpe LT, Nordby K, ed. Night vision: basic, clinical and applied aspects. Cambridge: Cambridge University Press, 1990:253–89.
- 7.Sharpe LT, Stockman A, Jagle H, et al. Opsin genes, cone photopigments amd colourblindness. In: Gegenfurtner K, Sharpe LT, ed. Color vision: from genes to perception. Cambridge: Cambridge University Press, 1999:3–52.
- 8.Wissinger B, Jägle H, Kohl S, et al. Human rod monochromacy: linkage analysis and mapping of a cone photoreceptor expressed candidate gene on chromosome 2q11. Genomics 1998;51:325–31. [DOI] [PubMed] [Google Scholar]
- 9.Kohl S, Marx T, Giddings I, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet 1998;19:257–9. [DOI] [PubMed] [Google Scholar]
- 10.Wissinger B, Gamer D, Jägle H, et al.CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet 2001;69:722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winick JD, Blundell ML, Galke BL, et al. Homozygosity mapping of the achromatopsia locus in the Pingelapese. Am J Hum Genet 1999;64:1679–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohl S, Baumann B, Broghammer M, et al. Mutations in the CNGB3 gene encoding the beta-subunit of the cone photoreceptor cGMP-gated channel are responsible for achromatopsia (ACHM3) linked to chromosome 8q21. Hum Mol Genet 2000;9:2107–16. [DOI] [PubMed] [Google Scholar]
- 13.Sundin OH, Yang J- M, Li Y, et al. Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet 2000;25:289–93. [DOI] [PubMed] [Google Scholar]
- 14.Mollon JD. ‘…aus dreyerley Arten von Membranen oder Molekülen’: George Palmer’s legacy. In: Cavonius CR, ed. Colour vision deficiencies XIII. Kluwer Academic Publishers, 1997:3–20.
- 15.Kohl S, Baumann B, Rosenberg T, et al. Mutations in the cone photoreceptor G-protein alpha-subunit gene GNAT2 in patients with achromatopsia. Am J Hum Genet 2002;71:422–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aligianis IA, Forshew T, Johnson S, et al. Mapping of a novel locus for achromatopsia (ACHM4) to 1p and identification of a germline mutation in the α subunit of cone transducin (GNAT2). J Med Genet 2002;39:656–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marmor MF, Zrenner E. Standard for clinical electroretinography (1994 update). Doc Ophthalmol 1995;89:199–210. [DOI] [PubMed] [Google Scholar]
- 18.Bach M, Hawlina M, Holder GE, et al. Standard for pattern electroretinography. International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol 2000;101:11–18. [DOI] [PubMed] [Google Scholar]
- 19.Marmor MF, Zrenner E. Standard for clinical electro-oculography. International Society for Clinical Electrophysiology of Vision. Arch Ophthalmol 1993;111:601–4. [DOI] [PubMed] [Google Scholar]
- 20.Arden G, Wolf J, Berninger T, et al. S-cone ERGs elicited by a simple technique in normals and in tritanopes. Vis Res 1999;39:641–50. [DOI] [PubMed] [Google Scholar]
- 21.Mollon JD, Astell S, Reffin JP. A Minimalist test of colour vision. In: Drum B, Moreland JD, Serra A, ed. Colour vision deficiencies XI. Kluwer Academic Publishers, 1991:59–67.
- 22.Mollon JD, Reffin JP. A computer-controlled colour vision test that combines the principles of Chibret and of Stilling. J Physiol 1989;414:5P. [Google Scholar]
- 23.Regan BC, Reffin JP, Mollon JD. Luminance noise and the rapid determination of discrimination ellipses in colour deficiency. Vis Res 1994;34:1279–99. [DOI] [PubMed] [Google Scholar]
- 24.Simunovic MP, Votruba M, Regan BC, Mollon JD. Colour discrimination ellipses in patients with dominant optic atrophy. Vis Res 1998;38:3413–20. [DOI] [PubMed] [Google Scholar]
- 25.Eksandh L, Kohl S, Wissinger B. Clinical features of achromatopsia in Swedish patients with defined genotypes. Ophthalmic Genet 2002;23:109–20. [DOI] [PubMed] [Google Scholar]
- 26.Stryer L. Visual excitation and recovery. J Biol Chem 1991;266:10711–14. [PubMed] [Google Scholar]
- 27.Morris TA, Fong SL. Characterisation of the gene encoding human cone transducin alpha-subunit (GNAT2). Genomics 1993;17:442–8. [DOI] [PubMed] [Google Scholar]
- 28.Fong SL. Characterisation of the human rod transducin alpha-subunit gene. Nucleic Acids Res 1992;20:2865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dryja TP, Hahn LB, Reboul T, et al. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet 1996;13:358–60. [DOI] [PubMed] [Google Scholar]
- 30.Cai K, Itoh Y, Khorana HG. Mapping of contact sites in complex formation between transducin and light-activated rhodopsin by covalent crosslinking: use of a photoactivatable reagent. Proc Natl Acad Sci 2001;98:4877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Arshavsky VY, Ruoho AE. Interaction sites of the COOH-terminal region of the gamma subunit of cGMP phosphodiesterase with the GTP-bound alpha subunit of transducin. J Biol Chem 1996;271:26900–7. [DOI] [PubMed] [Google Scholar]
- 32.Lerea CL, Bunt-Milam AH, Hurley JB. Alpha transducin is present in blue-, green-, and red-sensitive cone photoreceptors in the human retina. Neuron 1989;3:367–376. [DOI] [PubMed] [Google Scholar]
- 33.Reitner A, Sharpe LT, Zrenner E. Is colour vision possible with only rods and blue-sensitive cones? Nature 1991;352:798–800. [DOI] [PubMed] [Google Scholar]