

Abstract
Background/aims: To report the generation of a new mouse model for a genetically determined corneal abnormality that occurred in transgenesis experiments.
Methods: Transgenic mice expressing mutant forms of Rab27a, a GTPase that has been implicated in the pathogenesis of choroideremia, were generated.
Results: Only one transgenic line (T27aT15) exhibited an unexpected eye phenotype. T27aT15 mice developed corneal opacities, usually unilateral, and cataracts, resulting in some cases in phthisical eyes. Histologically, the corneal stroma was thickened and vacuolated, and both epithelium and endothelium were thinned. The posterior segment of the eye was also affected with abnormal pigmentation, vessel narrowing, and abnormal leakage of dye upon angiography but was histologically normal.
Conclusion: Eye abnormality in T27aT15 mice results from random insertional mutagenesis of the transgene as it was only observed in one line. The corneal lesion observed in T27aT15 mice most closely resembles posterior polymorphous corneal dystrophy and might result from the disruption of the equivalent mouse locus.
Keywords: corneal dystrophy, mutagenesis, transgenic mice
Corneal dystrophies form a group of rare and heterogeneous inherited eye disorders.1,2 They frequently develop during adolescence and progress gradually throughout life, but more severe cases are apparent at birth. Most dystrophies show an autosomal dominant pattern of transmission. These diseases are typically classified according to the layer(s) of the cornea primarily affected.2 Stromal dystrophies include lattice, granular, and macular dystrophy. Corneal dystrophies that affect the endothelium include Fuchs’ and posterior polymorphous dystrophy (PPCD).
Transgenesis has been a powerful methodology to study human disease processes. The use of transgenic disease models in biomedicine promises to dramatically accelerate the development of new human diagnostic and therapeutic treatments. To investigate the involvement of Rab27 in degenerative retinal diseases and other diseases where Rab27a might play a role, we generated transgenic mice overexpressing mutant forms of Rab27a.3 Most of the transgenic lines generated using this approach revealed the absence of any eye or other phenotype. However, one transgenic line exhibited an unexpected corneal phenotype. The phenotype in this line is probably not due to overexpression of Rab27a but rather to insertion of the transgene DNA into the genome. Transgene insertion is random and can be accompanied by other chromosomal rearrangements leading to the inactivation or activation of one or more endogenous genes. We report the phenotypic eye characterisation of this transgenic line as a possible animal model to study corneal dystrophy.
MATERIALS AND METHODS
Animals
All animals described here were maintained on 12 hour light/12 hour dark conditions at the CBS Unit, Imperial College London, under Home Office Project Licence 70/5056.
Generation of transgenic founders and lines
Transgenic mice expressing Rab27a dominant/negative (Rab27aT23N and Rab27aN133I) and constitutively active (Rab27aQ78L) mutant proteins under the control of the tyrosinase promoter were generated and characterised as described previously.3 Stable transgenic lines were propagated in the C57BL/6J background and maintained as hemizygous mice. Homozygous mice of this line were not produced because of poor breeding and early deaths of potentially homozygous mice.
Transgenic copy number determination
The number of copies integrated into the genome of transgenic mice was determined by Southern blotting as described previously.3 Briefly, 10 μg of transgenic mouse genomic DNA or wild type genomic DNA mixed with serially diluted microinjection DNA were digested with Pst I, separated on a 0.8% agarose gel, blotted onto Hybond-N+ membrane (Amersham Biosciences, Amersham, UK), hybridised with a 32P-labelled probe corresponding to the entire coding region of Rab27a, and then exposed to Hyperfilm MP (Amersham) in the presence of an intensifying screen or to a Multipurpose Screen followed by quantification using a Cyclotron Storage Phosphor Screen (Packard, Pangbourne, UK). Copy number was determined from a standard curve derived from the intensity of the signal in the spiked controls, assuming one copy of the transgene integration per diploid genome.
Fundus photography
Fundus photographs of mice were taken using a small animal fundus camera (Kowa Genesis, Tokyo, Japan).3,4 In order to have a better magnification and focal depth of the fundus, the camera was used in conjunction with an external 90 diopter condensing lens (Volk). Pupils were dilated with a drop of 1% Mydriacyl (Alcon Laboratories, Hemel Hempstead, UK) 20–30 minutes before taking the photographs. For conventional photography of the fundus, Kodak 200 ASA slide film was used. The photographic flash on the power pack was set up for its highest intensity of light (position 6–7).
Fluorescein angiography
The retinal angiography was performed using the general fundus photography procedure.3,4 The camera was set up for angiography through addition of an emission barrier filter specific for fluorescein emission, and the power pack was set up for fluorescein angiography by changing the excitation light to fluorescein excitation. For this type of photography Kodak black and white Tmax 400 ASA professional film was used. The photographic flash on the power pack was set at its highest level (position 6–7). Mice were injected intraperitoneally with 20% injectable sodium fluorescein (Faure, Annonay, France) at a dose of 10 μl per 5–6 g body weight.5 Photographs were taken at several intervals, starting at 30 seconds after injection.
Eye histology
Eyes were fixed in 4% paraformaldehyde, 5% glutaraldehyde, and 0.1 M cacodylate buffer for 1 hour. Subsequently, the eyes were cut longitudinally in half to separate the anterior and posterior segments. Fixed eyes were washed three times for 10 minutes at room temperature with PBS. Specimens were embedded in paraffin, sectioned to 3–5 μm thickness and stained with haematoxylin and eosin (to stain the nuclei and membranes, respectively) or periodic acid Schiff (PAS; to stain mucopolysaccharides from cornea).
RESULTS
Generation and characterisation of the T27aT15 transgenic line
Figure 1A provides a schematic diagram of the PTyr/Rab27aT23N/hGH expression construct.3 PTyr/Rab27aT23N/hGH includes a 2200 bp fragment of tyrosinase promoter,6 a 650 bp rat Rab27a coding sequence including the T23N point mutations, and 600 bp of sequence derived from the human growth factor gene which provides a polyadenylation signal. Figure 1B provides an example of screening for T27aT15 transgenic mice by Southern blotting. The analysis of the restriction digestion pattern revealed the presence of very strong bands around 3.4 kb, which probably corresponds to the expected bands for a tandem array of the transgene. The endogenous bands can hardly be detected in these Southern blots because of the strong signal derived from the transgenic bands. We estimate that the T27aT15 line contains approximately 260 copies of the transgene per cell, based on phosphor-imager quantification of the Southern blot signal (data not shown). We characterised four other independent lines containing the same construct for which we estimated the transgene copy number to range between 10–180 copies. The T27aT15 line probably contains a single integration site of the transgenes as there is no segregation of different restriction digestion patterns in the F1 generation.
Figure 1.

(A) Organisation of the PTyr/Rab27a/hGH transgenic construct. The point mutation in Rab27a is indicated by an asterisk. (B) Screening for potential transgenic mice by Southern blotting. Genomic DNA was extracted from the tail tips of offspring from a cross between a T27aT15 transgenic and a wild type mouse and subjected to Southern blotting as described under materials and methods. Mice 4 to 8 are transgenic. (C) RT-PCR quantification of transgenic mRNA. Total RNA was extracted from eyes of adult wild type, T27aT15 and T27aT17 transgenic mice and Rab27a mRNA was quantified and normalised to Hprt expression as described previously.3 (D) Expression of transgenic Rab27a proteins. Protein lysates (50 μg) from wild type, T27aT15, T27aT17, and A27aQ243 transgenic eyes were subjected to SDS-PAGE and analysed by immunoblotting using the monoclonal anti-Rab27a antibody 4B12 as described previously.3 Calnexin was used as a loading control and was detected using a specific polyclonal antibody.3
The T27aT15 transgenic line expresses approximately twice as much transgenic as endogenous Rab27a mRNA (fig 1C).3 Despite the high levels of mRNA, the transgenic protein levels were undetectable by immunoblot analysis (fig 1D). However, this result was observed for all other transgenic lines expressing the Rab27aT23N protein (see for example T27aT17 in fig 1D) and our results indicate that this is caused by instability of the mutant Rab27a protein.3
Phenotypic analysis of the anterior segment of the eye
About 20% of T27aT15 transgenic mice exhibited significant visual disorders. The most striking alterations of the anterior segment of the eye were corneal opacities and cataract. The opacity of the cornea, usually unilateral, became apparent at around 2 months of age and progressed slowly, extending centrally and circumferentially (fig 2). The opacities were not sex dependent and involved the visual axis in some cases resulting in reduced vision. In some of the mice, the corneal abormality led to a corneal abscess and then endophthalmitis, often associated with other defects such as cataract. This condition often evolved into phtisis bulbi (fig 2). Paraffin sections of the cornea stained with PAS revealed thinner epithelium with fewer cell layers (fig 3, labelled EP). Vacuoles were seen in the stroma, associated with a thickening of this layer (fig 3, labelled S). Vascularisation of the cornea was not observed. In advanced stages of the disease, the Descemet’s layer and the endothelium could also be affected (fig 3, labelled EN). However, a multilayered endothelium that is often characteristic of some corneal dystrophies was not observed. The abnormalities were progressive and by approximately six months of age, these corneal structures became completely disrupted (fig 3).
Figure 2.

External photographs of the eyes of T27aT15 transgenic mice. Eyes with an advanced abnormality of the cornea (A), a stromal opacity of the cornea (B), and a cataract (C) are shown. The arrow indicates a corneal abnormality area.
Figure 3.

Histochemical staining of T27aT15 mouse cornea. Transverse paraffin sections (5 μm) from T27aT15 transgenic mouse (B) and from a littermate control (A) were stained with periodic acid Schiff and visualised (magnification, ×130) as described under materials and methods. The abbreviations used are epithelium (EP), the avascular stroma (S) containing 200–300 parallel lamellae of collagen, and endothelium (EN). Bar = 300 μm.
Phenotypic analysis of the posterior segment of the eye
In most cases, observation of the eye fundus was hindered by the presence of corneal and lens opacities. We were nevertheless able to perform retinal analysis in those cases where anterior segment disease was less obvious. Photographic studies of retinal fundi of these transgenic mice revealed abnormal pigmentation around the optical nerve (fig 4). Furthermore, fluorescein fundus angiography revealed pathologic findings. Figure 5 depicts typical angiograms taken at different times after fluorescein dye injection and revealed abnormal leakage of dye from retinal capillaries. The arrows in figure 5 highlight an area with possible leakage of fluorescein dye. However, we do not exclude the possibility that this represents background fluorescence from the choriocapillaries shinning through the RPE. Alternatively, it could be caused by abnormal hyperpigmentation of the RPE or deficiency (lobular filling) of the choriocapillaries. We also noted that some mice presented with narrower blood vessels compared with non-transgenic mice (data not shown). This lesion seemed to be present only in those mice that developed serious anterior and posterior segment abnormalities and usually the eye became phthisical by six months of age. In addition, small hypopigmented areas that could result in transmission defects were observed in some of the animals affected. However, it is questionable whether the transmission defects were caused by loss of cells (usually RPE cells) or the pigmentation within cells. In contrast to what was observed for the cornea, the retina of these mice exhibited a normal appearance by histochemical analysis (fig 6).
Figure 4.

Fundus photographs of T27aT15 mice. Three month old wild type (A) and T27aT15 transgenic (B) mouse were examined as described under materials and methods. Hypopigmentation (a), a stromal opacity of the cornea (b), and a focal patch of RPE hyperpigmentation in the optical nerve area (c) are indicated. Note also the narrowing of retinal vessels.
Figure 5.

Fluorescein fundus angiograms of T27aT15 transgenic mouse retina. Angiograms were taken at 83, 141, 186, and 343 s after dye injection. The arrow indicates a leaking area.
Figure 6.
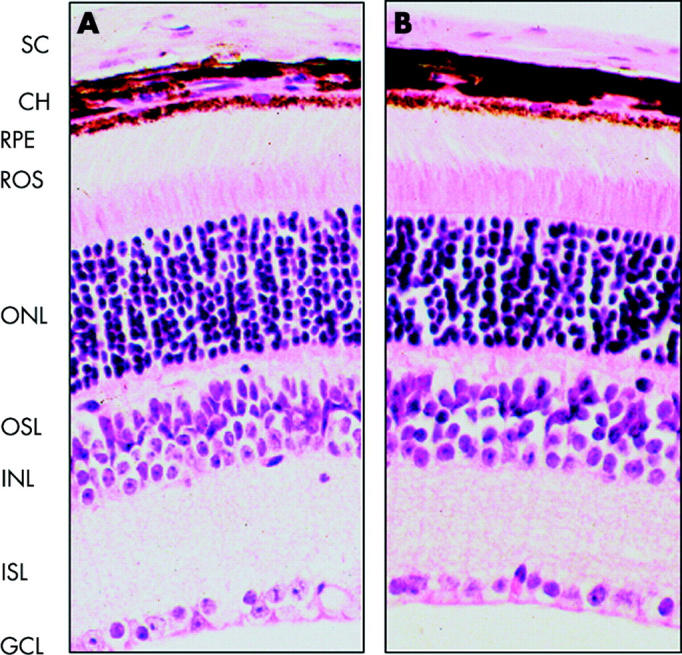
Histochemical staining of T27aT15 mouse retina. Transverse paraffin sections (3 μm) from 3 month old wild type (A) and T27aT15 (B) mice were stained with haematoxilin and eosin, and visualised (magnification, ×250) as described under materials and methods. SC, sclera; CH, choroids; RPE, retinal pigment epithelium; ROS, photoreceptor segments of rods and cones; ONL, nuclei of rods and cones; OSL, outer synaptic layer; INL, neuron nuclear layer; ISL, inner synaptic layer; GCL, ganglion cell layer.
DISCUSSION
In any transgenic mice, insertion of the transgene in the genome is potentially mutagenic. It has been proposed that about 5–10% of transgenic mice harbour transgene induced chromosomal alterations, including deletions and translocations.7,8 Thus, random insertion of foreign DNA into any genome is liable to deactivate or activate one or more genes. The striking eye phenotype observed in the T27aT15 transgenic line seems to be the result of the transgene insertion into the genome as the phenotype is unique among more than twenty transgenic lines that were generated using identical or similar constructs.3
We present a phenotypic characterisation of the T27aT15 line. A proportion of hemizygous transgenic mice developed serious abnormalities of the eye, usually starting between one and two months of age. The progressive eye abnormalities seem to arise after eye formation, as the eye appears to develop normally. The phenotype of these mice includes corneal opacity, cataract, and retinal vascular abnormalities. To date, we have not been able to find a description of mice with an inherited eye disease resembling that seen in these mice.
The phenotype observed in T27aT15 does not seem to be secondary to trauma inflicted upon the cornea, because if this was the case then the eyes would have been obviously inflamed as soon as the opacities were evident, there would have been tearing and discharge from the eye, and the mice would clearly have been distressed. None of these signs was evident. Thus, T27aT15 appears to be an example of a new type of corneal and eye dystrophy in the mouse.
Many different mouse genetic mutations result in cataract.9–12 However, there are only a few descriptions of mouse genetic corneal dystrophies.13–18 In the T27aT15 mice, the major and possibly primary defect is the dystrophy of the cornea. The abnormality of the corneal stroma, which includes the presence of vacuoles, could result from the heterozygous disruption or activation of a gene required for organisation and maintenance of this tissue. BIGH3 (transforming growth factor β induced gene) is a gene involved in corneal dystrophies such as lattice and granular corneal dystrophies characterised by deposits of amyloid or hyaline in the stroma.14,19,20 BIGH3 encodes an extracellular matrix adhesion protein (βig-h3) inducible by transforming growth factor β and abundant in the cornea.21 Although βig-h3 plays an important role in cornea physiology, its involvement in our mice is unlikely, given that the BIGH3 derived dystrophies are characterised by stromal deposits rather than vacuolisation of the stroma. Another possibility is that the transgenic insertional mutagenesis affected a regulatory gene, such as Pax6 or a similar gene. Pax6 is a master regulator of ocular morphogenesis and Pax6 mutations have been detected in various types of ocular anomalies, including aniridia, Peter’s anomaly, corneal dystrophy, congenital cataract, and foveal hypoplasia.17,22,23 The gene is expressed in the developing central nervous system, as well as in ocular tissues, including tissues of the corneal epithelium, lens, and retina. Pax6 is a transcription factor that recognises target genes through its paired type DNA binding domain.22,23
Another possibility that might explain the phenotype observed in T27aT15 mice is a multi-locus disruption by the transgene. Transgene integration is sometimes accompanied by alterations in the gross structure of the chromosome near the integration site, resulting in multiple gene inactivation events. A less likely possibility is multiple integrations of the transgene in more than one chromosome. In this case, there is usually segregation in successive generations of the chromosomes affected, and our observations indicate that the transgene is stably transmitted through the germ line.
One interesting candidate appears to be the posterior polymorphous corneal dystrophy locus (PPCD, MIM 122000). This type of dystrophy is characterised by the presence of vacuoles in the posterior part of the cornea that can easily be detected by ophthalmological examination and often by the presence of multilayered endothelium with characteristics of an epithelium.24 In T27aT15 mice, similar vacuoles are detected, although the endothelium appears monolayered (fig 3). One case of PPCD has been found to be caused by mutations in the type VIII collagen gene, COL8A2.25 However, another PPCD locus was mapped to 20p11.2–q11.2.24 Almost 100% of the transgenic mice of the T27aT15 line were agouti even after four or five generations of backcrossing to C57BL/6J mice. This suggests that the transgenic integration is near to the agouti locus, and that it integrated on the CBA derived chromosome in the CBA×C57BL/6J F2 embryo into which it was injected. Intriguingly the human homolog of the agouti locus, ASIP (agout mouse signaling protein, also known as AGTI), has been mapped to 20q11.2, near the PPCD locus (see www.ncbi.nlm.nih.gov/LocusLink/). Taken together, the similarities of corneal phenotype and human mapping position suggest that PPCD might be the locus disrupted.
Further studies will be required to better characterise the genotype and phenotype of these mice and to elucidate the relation between transgene integration and the phenotype observed. The mouse model described here represents a new model for genetic eye disease, and could be particularly relevant to the study of the pathogenesis of corneal dystrophy.
Acknowledgments
We thank Amanda McGuigan for making the transgenic mouse and Ross Anders for help with screening and handling of mice. This work was supported by the Medical Research Council, the Foundation Fighting Blindness, and an anonymous donor.
Abbreviations
PAS, periodic acid Schiff
PPCD, posterior polymorphous dystrophy
REFERENCES
- 1.Siddiqui N, Afshari NA. The changing face of the genetics of corneal dystrophies. Curr Opin Ophthalmol 2002;13:199–203. [DOI] [PubMed] [Google Scholar]
- 2.Arffa RC. Diseases of the Cornea. London: Mosby, 1991.
- 3.Ramalho JS, Anders R, Jaissle GB, et al. Rapid degradation of dominant–negative Rab27 proteins in vivo precludes their use in transgenic mouse models. BMC Cell Biol 2002;3:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawes NL, Smith RS, Chang B, et al. Mouse fundus photography and angiography: a catalogue of normal and mutant phenotypes. Mol Vis 1999;5:22. [PubMed] [Google Scholar]
- 5.Okamoto N, Tobe T, Hackett SF, et al. Transgenic mice with increased expression of vascular endothelial growth factor in the retina: a new model of intraretinal and subretinal neovascularization. Am J Pathol 1997;151:281–91. [PMC free article] [PubMed] [Google Scholar]
- 6.Beermann F, Schmid E, Schutz G. Expression of the mouse tyrosinase gene during embryonic development: recapitulation of the temporal regulation in transgenic mice. Proc Natl Acad Sci U S A 1992;89:2809–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meisler MH. Insertional mutation of ‘classical’ and novel genes in transgenic mice. Trends Genet 1992;8:341–4. [DOI] [PubMed] [Google Scholar]
- 8.Rijkers T, Peetz A, Ruther U. Insertional mutagenesis in transgenic mice. Transgenic Res 1994;3:203–15. [DOI] [PubMed] [Google Scholar]
- 9.Bloemendal H. Proctor lecture. Disorganization of membranes and abnormal intermediate filament assembly lead to cataract. Invest Ophthalmol Vis Sci 1991;32:445–55. [PubMed] [Google Scholar]
- 10.Gotz W. Transgenic models for eye malformations. Ophthalmic Genet 1995;16:85–104. [DOI] [PubMed] [Google Scholar]
- 11.Graw J. Mouse models of congenital cataract. Eye 1999;13:438–44. [DOI] [PubMed] [Google Scholar]
- 12.Favor J, Neuhauser-Klaus A. Saturation mutagenesis for dominant eye morphological defects in the mouse Mus musculus. Mamm Genome 2000;11:520–5. [DOI] [PubMed] [Google Scholar]
- 13.Irvine AD, Corden LD, Swensson O, et al. Mutations in cornea-specific keratin K3 or K12 genes cause Meesmann’s corneal dystrophy. Nat Genet 1997;16:184–7. [DOI] [PubMed] [Google Scholar]
- 14.Munier FL, Korvatska E, Djemai A, et al. Kerato-epithelin mutations in four 5q31-linked corneal dystrophies. Nat Genet 1997;15:247–51. [DOI] [PubMed] [Google Scholar]
- 15.Tsujikawa M, Kurahashi H, Tanaka T, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nat Genet 1999;21:420–3. [DOI] [PubMed] [Google Scholar]
- 16.Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nat Genet 2000;26:237–41. [DOI] [PubMed] [Google Scholar]
- 17.Glaser T, Jepeal L, Edwards JG, et al. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet 1994;7:463–71. [DOI] [PubMed] [Google Scholar]
- 18.Chakravarti S, Petroll WM, Hassell JR, et al. Corneal opacity in lumican-null mice: defects in collagen fibril structure and packing in the posterior stroma. Invest Ophthalmol Vis Sci 2000;41:3365–73. [PMC free article] [PubMed] [Google Scholar]
- 19.Stewart HS, Ridgway AE, Dixon MJ, et al. Heterogeneity in granular corneal dystrophy: identification of three causative mutations in the TGFBI (BIGH3) gene-lessons for corneal amyloidogenesis. Hum Mutat 1999;14:126–32. [DOI] [PubMed] [Google Scholar]
- 20.Schmitt-Bernard CF, Guittard C, Arnaud B, et al. BIGH3 exon 14 mutations lead to intermediate type I/IIIA of lattice corneal dystrophies. Invest Ophthalmol Vis Sci 2000;41:1302–8. [PubMed] [Google Scholar]
- 21.Skonier J, Neubauer M, Madisen L, et al. cDNA cloning and sequence analysis of βig-h3, a novel gene induced in a human adenocarcinoma cell line after treatment with transforming growth factor-β. DNA Cell Biol 1992;11:511–22. [DOI] [PubMed] [Google Scholar]
- 22.Ashery-Padan R, Gruss P. Pax6 lights-up the way for eye development. Curr Opin Cell Biol 2001;13:706–14. [DOI] [PubMed] [Google Scholar]
- 23.Gehring WJ. The genetic control of eye development and its implications for the evolution of the various eye-types. Int J Dev Biol 2002;46:65–73. [PubMed] [Google Scholar]
- 24.Heon E, Mathers WD, Alward WL, et al. Linkage of posterior polymorphous corneal dystrophy to 20q11. Hum Mol Genet 1995;4:485–8. [DOI] [PubMed] [Google Scholar]
- 25.Biswas S, Munier FL, Yardley J, et al. Missense mutations in COL8A2, the gene encoding the alpha2 chain of type VIII collagen, cause two forms of corneal endothelial dystrophy. Hum Mol Genet 2001;10:2415–23. [DOI] [PubMed] [Google Scholar]