

Abstract
Background/aims: To present the clinical, psychophysical, and electrophysiological characteristics of a family with dominantly inherited congenital stationary night blindness (CSNB).
Methods: Five affected family members from three generations were ascertained. Four affected individuals underwent ophthalmic examination and electrodiagnostic investigations. Three affected individuals also underwent scanning laser ophthalmoscopy and psychophysical testing.
Results: Affected individuals reported night blindness from an early age. Visual acuities were normal. Fundal appearances were normal apart from one older patient showing areas of peripheral chorioretinal atrophy. Autofluorescence images showed no gross abnormality. International Society for Clinical Electrophysiology of Vision (ISCEV) standard electroretinography (ERG) showed undetectable rod specific responses and electronegative maximal responses, but normal ISCEV cone responses. Additional S-cone specific ERG recordings were of reduced amplitude in all patients studied. There was no apparent rod component to the dark adaptation curve. Central 30° thresholds were normal under photopic conditions but showed increased thresholds under scotopic conditons for both red and blue stimuli.
Conclusion: Results from investigation of this family are consistent with an impairment of rod photoreceptor signalling. The ERG findings suggest an abnormality occurring after phototransduction with rod and S-cone pathway involvement. These findings differ from those rare families reported previously with dominant CSNB.
Keywords: congenital stationary night blindness
Congenital stationary night blindness (CSNB) refers to a group of disorders characterised by infantile onset of nyctalopia and non-progressive retinal dysfunction that can be inherited as X linked recessive, autosomal recessive, or autosomal dominant traits. Autosomal dominant CSNB occurs in the Nougaret family as described by Cunier and Nettleship,1,2 and the psychophysical and electrophysiological findings have been described more recently.3 X linked and recessive CSNB is associated with a negative electroretinography (ERG) in the maximal response, where there is selective loss of the b-wave,4 termed as “Schubert-Bornschein” type. Miyake subdivides the Schubert-Bornschein type of CSNB into “complete” or “incomplete” according to the degree of rod function.5 Although originally based on electrophysiological and psychophysical criteria, these have subsequently been shown to reflect genetically distinct disorders.6–9 Patients with “complete” X linked CSNB have less obvious cone ERG abnormalities than “incomplete” CSNB. There have been very few previous reports of negative ERG in dominant CSNB.10–12
We present the clinical, psychophysical, and electrophysiological characteristics of a new family with dominantly inherited congenital stationary night blindness (CSNB).
PATIENTS AND METHODS
Four affected individuals underwent ophthalmic examination including Snellen acuity and biomicroscopic fundoscopy. Retinal appearance was documented with colour photography and one patient (subject III-2) underwent fluorescein angiography.
ELECTROPHYSIOLOGY
Subjects underwent electrophysiological investigation using techniques in accordance with the recommendations of the International Society for Clinical Electrophysiology of Vision.13–15 Electro-oculographic responses (EOG), full field ERG, and pattern electroretinograms (PERG) were recorded. ON and OFF responses were recorded using a mini Ganzfeld stimulator (CH Electronics, Bromley, UK), based on light emitting diode technology, using a 200 ms, 530 nm stimulus of 440 candelas (cd) per square metre, on a background of 612 nm and 160 cd/m2, and a stimulus rate of two per second.16 S-cone specific ERGs were recorded using a 5 ms blue stimulus on an amber background as previously described.17
PSYCHOPHYSICAL TESTS
Static threshold perimetry in the dark and light adapted states was performed using a Humphrey field analyser (Allergan Humphrey, Hertford, UK). Photopic visual fields were performed using the standard protocol. For dark adapted visual fields, the pupil was dilated with 2.5% phenylephrine hydrochloride and 1% cyclopentolate hydrochloride, and the patient dark adapted for 45 minutes. The Humphrey field analyser was modified for use in dark adapted conditions.18–20 An infrared source illuminated the bowl, and an infrared monitor (Philips, Eindhoven, Holland) was used to detect eye movements. Fields were recorded using the central 30-2 threshold test. The target size corresponded to Goldmann size V. Each programme was performed with a red (dominant wavelength, 650 nm) and then blue (dominant wavelength, 450 nm) filter in the stimulus beam. The dark adapted blue central 30-2 fields were reviewed to determine the most informative locations of dark adapted visual sensitivity. Two test locations were chosen at 3° and 9°. The Humphrey field analyser was used for dark adaptometry controlled by a customised computer program (PS/2 model 50; International Business Machines, Armonk, NY, USA).20,21 Fully dark adapted rod thresholds were measured before exposure to the adapting light at the two locations with the blue filter in the stimulus beam.
AUTOFLUORESCENCE IMAGING
Confocal scanning laser ophthalmoscope images of the central macular region were obtained using a prototype cLSO SM 30-4024, (Zeiss, Oberkochen, Germany). An argon laser (488 nm, 250 µW) was used for illumination. Reflectance imaging was undertaken using the Zeiss LSO with a 40° field and the argon blue laser, with the depth plane adjusted to maximise the visibility of the fundus features. A wide bandpass filter, with a cut off at 521 nm inserted in front of the detector, was used to detect autofluorescence arising from lipofuscin at the level of the retinal pigment epithelium, which was recorded using published techniques.22
CASES AND RESULTS
Four of five affected individuals were examined and investigated (fig 1):
Figure 1.

Pedigree of this family. Roman numerals indicate the five generations ascertained. Solid symbols represent affected members and open symbols represent unaffected family members. Squares, males; circles, females; diagonal lines, deceased; vertical line, affected by hearsay.
Case 1 (III:2)
A 58 year old female presented with a history of nyctalopia since childhood. Best corrected visual acuity was 6/5 in each eye. Fundoscopy was normal apart from small areas of retinal pigment epithelium atrophy in both maculae and larger areas of chorioretinal atrophy in the periphery (fig 2). Fluorescein angiography showed small “window defects” in both maculae and larger areas in the periphery that corresponded to the clinical retinal appearance (fig 2). SLO autofluorescence imaging was normal apart from the atrophic areas (fig 3).
Figure 2.

Colour photographs of the fundus of the first case (III:2) illustrating the abnormal appearance of the macula and the peripheral retina (case 1). Fluorescein angiography of the first case showed small areas of hyperfluorescence at the central macula and larger areas in the periphery that correspond to the clinically observed chorioretinal atrophic patches (case 1). Fundus photographs of the third (IV:3) case were normal bilaterally (case 3).
Figure 3.
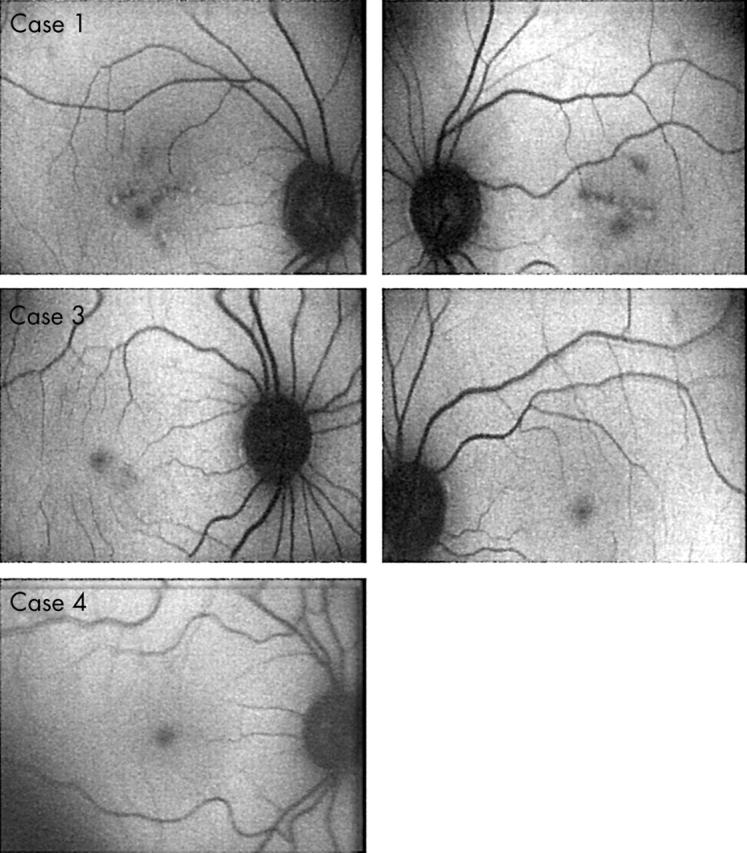
A prototype Zeiss confocal using argon laser blue light and a broadband pass barrier filter with a short wavelength cut off at 521 nm. Scanning laser ophthalmoscope autofluorescence imaging of the retina showing the abnormal areas in first case (III:2). Cases 3 and 4 autofluorescence imaging was normal.
Case 2 (IV:2)
The 38 year old son of case 1 also reported non-progressive night blindness since childhood. Unaided visual acuity was 6/5 in the right eye and 6/6 in the left. Fundal examination was normal.
Case 3 (IV:3)
The 36 year old daughter of case 1 also reported non-progressive night blindness since childhood. Unaided visual acuity was 6/5 bilaterally. Fundoscopic examination was normal (fig 2). SLO autofluorescence imaging was normal (fig 3).
Case 4 (V:1)
The 13 year old grandson of patient 1 also reported night blindness. Visual acuities and fundal appearance were normal. SLO autofluorescence imaging was normal (fig 3).
All four patients examined showed normal International Society for Clinical Electrophysiology of Vision (ISCEV) standard cone ERGs (the lower limit of normal photopic b-wave amplitude is 90 μV). S-cone ERGs were reduced in all tested subjects (the S-cone component at 50 ms should be of higher amplitude than the L/M-cone component at 30 ms when blue stimulation is used). The rod specific response was undetectable in all four patients. The maximal responses were markedly “electronegative”, with patients 1, 2, and 3 also showing mild a-wave amplitude abnormality in the “standard” bright white flash dark adapted response. Photopic ON and OFF responses were normal (fig 4).
Figure 4.

Full field electroretinography (ERG) and pattern ERG were recorded using standardised methods according to ISCEV standards. Long duration photopic stimulation was performed to separate ON and OFF photopic pathways. Investigations from a normal subject and a male affected with complete X linked congenital stationary night blindness are shown for comparison.
Central 30° photopic visual fields demonstrated sensitivities within normal range values throughout the visual field for all three tested subjects (fig 5).
Figure 5.

(A) Central 30-2 static threshold perimetry to light and dark adapted states (using red and blue stimulus) showing mean thresholds for 14 normal subjects. (B, C, and D) Dark adapted scotopic red and blue 30° static perimetry showing sensitivity loss in all three patients. Recorded photopic function was within normal range.
An overall depression of sensitivity was observed during scotopic testing with red and blue stimuli in all patients, with the loss of sensitivity being more severe with the blue stimulus for all subjects. The extent of central sensitivity loss in the scotopic blue perimetry was greater than 35 dB in all subjects. No specific pattern of sensitivity loss was detected. It is of interest that the youngest subject seems to be more severely affected, as shown in his dark adapted perimetry to red stimuli. In the scotopic blue perimetry the younger patient showed less sensitivity loss than the older patient but more sensitivity loss than case C (fig 5). It is also evident that there is lack of progression of sensitivity loss with age.
Formal dark adaptometry was performed in cases 3 and 4 and showed a monophasic curve with an absent rod component with normal final cone threshold (fig 6).
Figure 6.
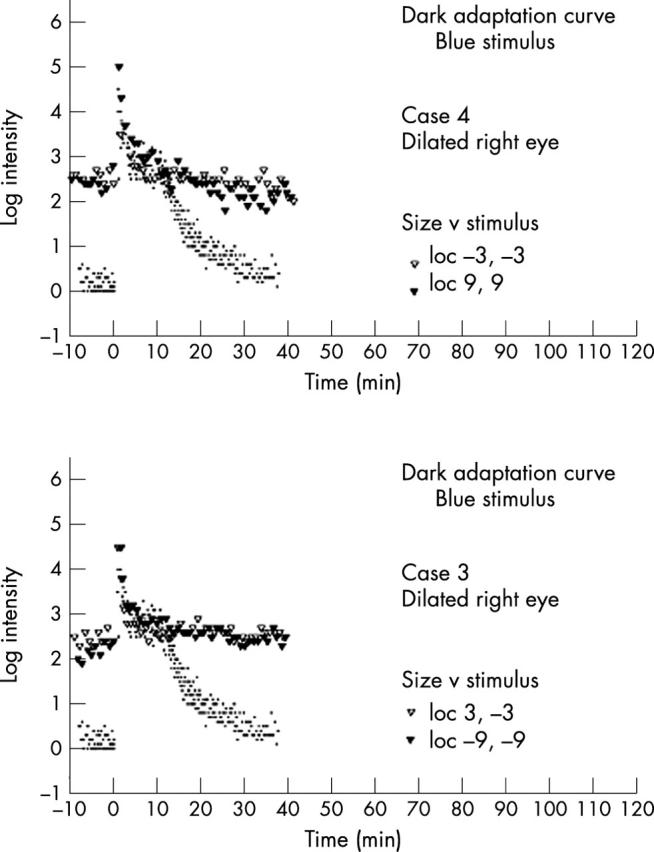
Dark adaptation curves. Log threshold using red test flash following strong adaptation plotted against time in the dark as measured at 3° and 9° from centre. Dots represent normal controls from averaging of three normal patients under similar testing conditions.
DISCUSSION
We describe a novel phenotype of congenital stationary night blindness with autosomal dominant inheritance and a Schubert-Bornschein type negative ERG. Affected individuals in this family reported night blindness from an early age with normal visual acuities. There was no significant refractive error in any affected family member. Fundal appearances were normal apart from the eldest patient who had small areas of retinal pigment epithelial atrophy in both maculae and larger areas of peripheral chorioretinal atrophy, which may be an unrelated finding.
All four patients examined showed a negative ERG, but no abnormality in standard cone ERGs, pattern ERG (which reflects macular function), or photopic ON and OFF responses, which arise in relation to L-cone and M-cone pathways. In addition, there was a consistent loss of S-cone ERGs. This family differs from those previously reported in that although a negative ERG was a consistent finding, in older patients there was a mild maximal response a-wave abnormality, suggesting some loss of rod photoreceptor function. Overall, the ERGs suggest predominant post-phototransduction dysfunction in rod and S-cone systems.
Autosomal dominant CSNB has been reported in a large French pedigree, the Nougaret family, and traced through 11 generations. Initial ERG findings in affected patients showed almost complete lack of rod function but normal cone responses.23 More recent electrodiagnostic investigation in two affected descendants (father and son) of the Nougaret family showed residual rod function with mild impairment of cone function.3 In the same study, dark adaptation measured in the son showed a possible rod-cone break suggestive of residual rod function. The family reported in the present study differs from the Nougaret family3 in that cone function in our cases was normal apart from the S-cone system, and the dark adaptation curve was monophasic for both examined members.
Other autosomal dominant CSNB families have been reported. Noble et al10 reported a family which showed a normal a-wave in the maximal full field ERG but otherwise resembled the family reported here. Those authors did not report PERG, ON/OFF response, or S-cone ERG data. A Japanese family with autosomal dominant CSNB has been reported11 but again showed a normal maximal a-wave response. The rod specific ERG was undetectable in all members of our family in contrast with the Japanese family where it was only diminished in one patient. A biphasic dark adaptation curve was also shown in one affected member of the Japanese family.
Specific mutations in the rhodopsin gene24–26 and in the cGMP phosphodiesterase beta subunit gene27 have been shown to cause autosomal dominant CSNB. A mutation (Gly38Asp) in the alpha subunit of rod transducin has been shown to cause the disorder in descendents of the original Nougaret family.28 Two X chromosomal loci have been identified for X linked CSNB, and the causative genes identified.6–9,29 Future genetic analysis of our family for candidate genes may shed light on the molecular pathogenesis of this ERG phenotype and on rod and S-cone signalling in the mammalian retina.
Abbreviations
CSNB, congenital stationary night blindness
EOG, electro-oculography
ERG, electroretinography
ISCEV, International Society for Clinical Electrophysiology of Vision
PERG, pattern electroretinogram
SLO, scanning laser ophthalmoscopy
REFERENCES
- 1.Cunier F . Histoire d’une hemeralopie hereditaire depuis deux siecles dans une famille de la commune de Vendemian pres de Montpelier. Ann Soc Med Gand 1838;4:385–95. [Google Scholar]
- 2.Nettleship E . A pedigree of congenital night blindness with myopia. Trans Ophthal Soc UK 1912;32:21–45. [Google Scholar]
- 3.Sandberg MA, Pawlyk BS, Dan J, et al. Rod and cone function in the Nougaret form of stationary night blindness. Arch Ophthalmol 1998;116:867–72. [DOI] [PubMed] [Google Scholar]
- 4.Schubert G , Bornschein H. Beitrag zur Analyse des menschlichen Electroretinogram. Ophthalmologica 1952;123:396–413. [DOI] [PubMed] [Google Scholar]
- 5.Miyake Y , Yagasaki K, Horiguchi M, et al. Congenital stationary night blindness with negative electroretinogram. A new classification. Arch Ophthalmol 1986;104:1013–20. [DOI] [PubMed] [Google Scholar]
- 6.Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet 1998;19:264–7. [DOI] [PubMed] [Google Scholar]
- 7.Strom TM, Nyakatura G, Apfelstedt-Sylla E, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet 1998;19:260–3. [DOI] [PubMed] [Google Scholar]
- 8.Bech-Hansen NT, Naylor MJ, Maybaum TA, et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet 2000;26:319–23. [DOI] [PubMed] [Google Scholar]
- 9.Pusch CM, Zeitz C, Brandau O, et al. The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat Genet 2000;26:324–7. [DOI] [PubMed] [Google Scholar]
- 10.Noble KG, Carr RE, Siegel IM. Autosomal dominant congenital stationary night blindness and normal fundus with an electronegative electroretinogram. Am J Ophthalmol 1990;109:44–8. [DOI] [PubMed] [Google Scholar]
- 11.Hayakawa M , Imai Y, Wakita M, et al. A Japanese pedigree of autosomal dominant congenital stationary night blindness with variable expressivity. Ophthalmic Paediatr Genet 1992;13:211–17. [DOI] [PubMed] [Google Scholar]
- 12.Sverak J , Jebava R, Peregrin J, et al. [Congenital stationary night blindness]. Cesk Slov Oftalmol 1996;52:135–42. [PubMed] [Google Scholar]
- 13.Marmor MF, Zrenner E. Standard for clinical electroretinography (1994 update). Doc Ophthalmol 1995;89:199–210. [DOI] [PubMed] [Google Scholar]
- 14.Marmor MF, Holder GE, Porciatti V, et al. Guidelines for basic pattern electroretinography. Recommendations by the International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol 1995;91:291–8. [DOI] [PubMed] [Google Scholar]
- 15.Marmor MF, Zrenner E. Standard for clinical electro-oculography. International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol 1993;85:115–24. [DOI] [PubMed] [Google Scholar]
- 16.Downes SM, Holder GE, Fitzke FW, et al. Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch Ophthalmol 2001;119:96–105. [PubMed] [Google Scholar]
- 17.Arden G , Wolf J, Berninger T, et al. S-cone ERGs elicited by a simple technique in normals and in tritanopes. Vision Res 1999;39:641–50. [DOI] [PubMed] [Google Scholar]
- 18.Jacobson SG, Voigt WJ, Parel JM, et al. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology 1986;93:1604–11. [DOI] [PubMed] [Google Scholar]
- 19.Steinmetz RL, Haimovici R, Jubb C, et al. Symptomatic abnormalities of dark adaptation in patients with age-related Bruch’s membrane change. Br J Ophthalmol 1993;77:549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alexander KR, Fishman GA. Prolonged rod dark adaptation in retinitis pigmentosa. Br J Ophthalmol 1984;68:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JC, Fitzke FW, Pauleikhoff D, et al. Functional loss in age-related Bruch’s membrane change with choroidal perfusion defect. Invest Ophthalmol Vis Sci 1992;33:334–40. [PubMed] [Google Scholar]
- 22.von Ruckmann A , Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol 1995;79:407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Francois J , Verriest G, de Rouck A, et al. Les fonctions visuelles dans l’hemeralopie essentielle nougarienne. Ophthalmologica 1956;132:244–57. [DOI] [PubMed] [Google Scholar]
- 24.Dryja TP, Berson EL, Rao VR, et al. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat Genet 1993;4:280–3. [DOI] [PubMed] [Google Scholar]
- 25.Al-Jandal N , Farrar GJ, Kiang AS, et al. A novel mutation within the rhodopsin gene (Thr-94-Ile) causing autosomal dominant congenital stationary night blindness. Hum Mutat 1999;13:75–81. [DOI] [PubMed] [Google Scholar]
- 26.Sieving PA, Richards JE, Naarendorp F, et al. Dark-light: model for nightblindness from the human rhodopsin Gly90→Asp mutation. Proc Natl Acad Sci USA 1995;92:880–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gal A , Orth U, Baehr W, et al. Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat Genet 1994;7:551. [DOI] [PubMed] [Google Scholar]
- 28.Dryja TP, Hahn LB, Reboul T, et al. Missense mutation in the gene encoding the alpha subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet 1996;13:358–60. [DOI] [PubMed] [Google Scholar]
- 29.Zito I , Allen LE, Patel RJ, et al. Mutations in the CACNA1F and NYX genes in British CSNBX families. Hum Mutat 2003;21:169. [DOI] [PubMed] [Google Scholar]