

Abstract
Aim: To describe the clinical characteristics and disease course of a large family with retinitis pigmentosa (RP) from an Arg135Leu change in rhodopsin.
Methods: 29 patients in this family were evaluated. Goldmann visual fields were performed on 14 affected individuals, Ganzfeld electroretinography (ERG) on eight individuals (11–56 years), and blood samples collected on 10 individuals (11–58 years). Patient visual field data were compared with previously reported patients with different rhodopsin mutations using linear regression.
Results: An Arg135Leu mutation was identified in rhodopsin. Distinct stages of clinical evolution were identified for this family ranging from normal, white dots, classic bone spicules and, finally, ending with extensive retinal pigment epithelium (RPE) atrophy. 9/16 patients over the age of 20 years also demonstrated marked macular atrophy. All patients who underwent full field ERG testing demonstrated non-recordable ERGs. The overall regression model comparing solid angles of visual fields from patients with rhodopsin mutations (Pro23His, Pro347Ala, Arg135Leu) shows significant effects for age (p = 0.0005), mutation (p = 0.0014), and interaction between age and mutation (p = 0.018) with an R2 of 0.407.
Conclusions: An Arg135Leu change in rhodopsin results in a severe form of RP that evolves through various fundus appearances that include white dots early in life and classic appearing RP later. This transmembrane change in rhodopsin proves to be more severe than in a family with an intradiscal change and a family with a cytoplasmic change.
Keywords: multifocal electroretinography, retinitis pigmentosa, retinal dystrophy, retinitis punctata albescens, rhodopsin
Retinitis pigmentosa (RP) is characterised by clinical findings of bone spicules, arteriolar attenuation, and optic nerve atrophy. However, there is a wide range of presentation and severity. The clinical appearance of any given patient with RP depends on the stage of the patient in the disorder and the molecular basis of disease. Of the approximately 100 000 patients with RP in the United States, 15–20% of patients have autosomal dominant retinitis pigmentosa (ADRP).1–3 The most common molecular causes of ADRP are changes in rhodopsin. Rhodopsin related RP tends to have a milder disease course than other forms of RP.1,4 However, Sandberg demonstrated that the severity of rhodopsin related RP was dependent on the site of the sequence variation.5 An Arg135Leu mutation in rhodopsin was reported by Sung.4,6,7 We describe the clinical characteristics of this mutation in a 13 generation family. This change in rhodopsin demonstrates a very severe course with widespread white dots early that fade into a more classic appearance of RP in adulthood.
SUBJECTS AND METHODS
This study had approval of the institutional review board of the University of North Carolina (UNC). A total of 24 affected patients, all members of a 13 generation family with ADRP (fig 1), were examined at UNC. Five additional affected and multiple unaffected patients were examined at a family reunion by the author. Patients clearly reported nyctalopia from early childhood and this symptom was used to determine disease status. Affected patients from four different generations and two separate branches of the family came to UNC. Patients underwent a standard eye examination, Goldmann perimetry, electroretinographic testing, and fundus photography. Patients with hand movement or worse vision did not undergo ancillary testing. Full field electroretinography and multifocal electroretinography (mfERG) were performed according to ISCEV standards.8,9 Eight patients underwent full field ERG testing with an LKC Veritas system (Gaithersburg, MD, USA). Three patients underwent mfERG using a Veris system (ElectroDiagnostics, Inc, San Mateo, CA, USA). Goldmann visual fields (GVF) were converted to solid angle (in steradians) using described methods.10,11 A linear regression model was performed from patients with Arg135Leu, Pro23His and Pro347Ala mutations in rhodopsin11 (JMP SAS, Cary, NC, USA).
Figure 1.

Pedigree. The family, which clearly demonstrates an autosomal dominant pattern of inheritance, could be traced back 13 generations. The branches of the family that participated in the current study are boxed. From the left box, one woman from generation X, five patients from generation XI, and two patients from generation XII were examined. From the right box, seven patients from generation XI, seven patients from generation XII, and two patients from generation XIII were examined at UNC. The majority of living affected and unaffected members of these two branches of the family were examined at the family reunion in western North Carolina.
Molecular analysis and findings
Ten patients at various disease stages ranging in age from 11–58 years had blood samples sent to the Carver Molecular Ophthalmology Laboratory for analysis. Screening using an amplification refractory mutation system (ARMS)12 disclosed neither the Pro23His nor Arg135Trp changes in rhodopsin. Samples that had aberrant migration patterns of polymerase chain reaction products, using single stranded conformational polymorphism analysis,11,13,14 were sequenced bidirectionally with fluorescent dideoxynucleotides on an ABI model 377 automated sequencer. An Arg135Leu change in rhodopsin was identified in all samples.
RESULTS
Clinical findings
All affected patients in this study, ranging in age from 7–64 years of age, reported symptoms of nyctalopia before 5 years of age. No unaffected patients demonstrated white dots or other signs of RP. Visual acuity ranged from 20/20 to light perception and remained stable until the development of posterior subcapsular cataracts in the third to fourth decade of life (fig 2). Nearly all patients over the age of 16 who were evaluated had cataracts or intraocular lenses with maintained central visual acuity until the fourth to fifth decade of life. However, 9/16 patients over the age of 20 years demonstrated some degree of macular atrophy that compromised central vision (fig 3A). Six of seven patients under the age of 20 years who were examined, demonstrated widespread white dots and minimal RPE changes with only the youngest patient (age 7 years) not demonstrating any definite signs (fig 3B and 3C). In the second to third decades, the white dots fade and are replaced by RPE atrophy and bone spicules (fig 3D) and, finally, the fundus shows marked RPE atrophy and choroidal sclerosis (fig 3E and F).
Figure 2.

Visual acuity. The central visual acuity of the better seeing eye of each patient is plotted against their age at examination. From this distribution, there is a notable drop off in central visual acuity by the fourth decade of life. Outliers who maintain central visual acuity beyond the fourth decade of life notably have minimal macular atrophy.
Figure 3.
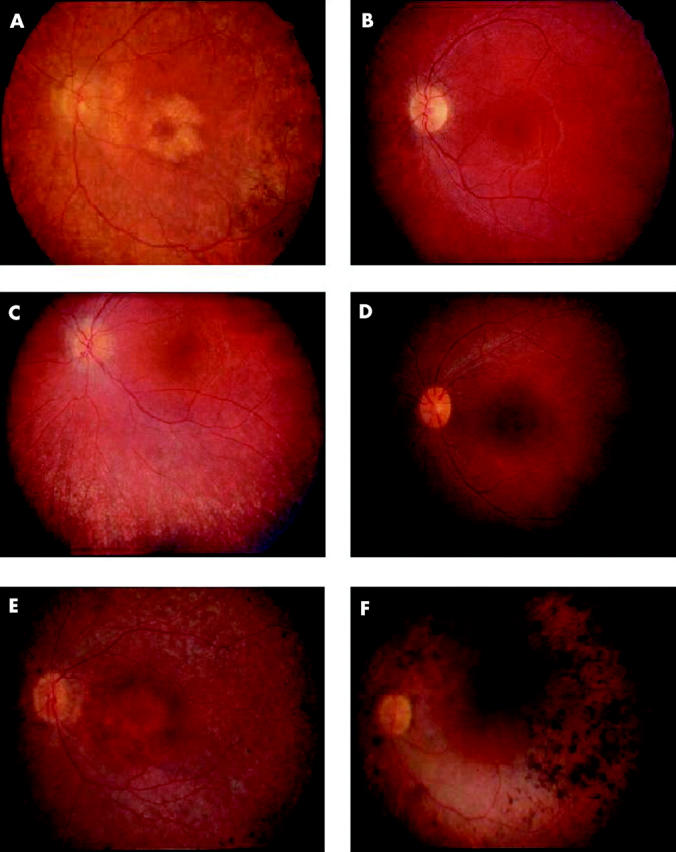
Fundus appearance. (A) Fundus image of affected man, aged 41 years. Note the parafoveal atrophy of the retinal pigment epithelium (RPE). There is RPE atrophy and bone spicules beyond the arcades. Arterioles are attenuated. (B) Fundus image of affected boy, aged 9 years. Note the relative lack of fundus findings. This patient has some pallor to the optic nerve and attenuation of his arterioles. Periphery demonstrated no evidence of RPE atrophy, bone spicules, or white dots. (C) Fundus image of affected girl, aged 11. Note the extensive white dots and RPE mottling in the mid-periphery. Bone spicules are notably absent. (D) Fundus image of affected girl, aged 16. Note the presence of white dots and RPE atrophy. Bone spicules are beginning to develop in the mid-periphery. (E) Fundus image of affected woman, aged 45 years. Note the extensive RPE thinning and atrophy both in the mid-periphery and the macula. The optic nerve is pale and there is arteriolar attenuation. (F) Fundus image of affected man, aged 57 years. Note the extensive RPE clumps in the mid-periphery and visible sclerotic choroidal vessels.
Electroretinographic findings
We evaluated eight patients with full field electroretinography (ages 11–50 years) and three patients with mfERG. All patients undergoing full field electroretinography showed non-recordable responses. For mfERG testing, only the 16 year old demonstrated recordable retinal function, though no other patients younger than 40 years were tested. Repeat mfERG for this patient 13 months later found considerable further loss of recordable amplitude.
Perimetric findings
Fifteen patients underwent Goldmann perimetry. There was no measurable field with the I4e isoptre by the fourth decade of life. The V4e test target became undetectable by the sixth decade of life (fig 4). When the solid angle of I4e isoptre was compared to the clinical characteristics of the Pro23His and the Pro347Ala change in rhodopsin, there was a significant difference in the loss of visual field for patients with an Arg135Leu change in rhodopsin compared with the other mutations. The difference between visual field data for patients with the rhodopsin Arg135Leu RP and those with either other change remained significant at the 0.0005 level after Bonferroni correction (fig 5). The overall regression model comparing patients with rhodopsin mutations (Pro23His, Pro347Ala, Arg135Leu) shows significant effects for age (p = 0.0005), mutation (p = 0.0014), and interaction between age and mutation (p = 0.018) with an R2 of 0.407. Comparison of Arg135Leu change and Pro23His change demonstrated a significant difference in these populations (p = 0.0002) but no significance in the effect of age or the interaction of age and mutation. Comparison of Arg135Leu change and Pro347Ala change shows significant effect of age (p = 0.0001), mutation (p = 0.0001), and the interaction of age with mutation (p = 0.0003).
Figure 4.

Graph showing solid angle of visual field for patients for II4e and V4e isoptres at their particular age. II4e isoptres are depicted by diamonds and the V4e isoptres are depicted by squares. Note that the II4e isoptre is essentially unrecordable in patients over the age of 35 years and the V4e isoptre is minimally recordable by 50 years of age.
Figure 5.

Graph comparing the solid angle of visual field for the I4e isoptre of three mutations in rhodopsin: Pro23His, Pro347Ala, and Arg135Leu. Note that patients with the intradiscal change (Pro23His) have the most recordable visual field over time and that those with the transmembrane change (Arg135Leu) have the worst overall visual fields.
DISCUSSION
Rhodopsin related disease may present with a wide spectrum of severity and clinical features, including regional and diffuse RP,15–17 sectoral RP,18–23 and congenital stationary night blindness.24–27 Sandberg previously identified the importance of the locus of mutations in rhodopsin to a specific patient’s disease course, showing that intradiscal variations such as Pro23His carried the mildest course of disease and cytoplasmic variations such as at codon 347 carried the most severe prognosis.5,28 The severity of mutations in the intracytoplasmic region of rhodopsin is well documented though only a few clinical studies exist for changes in the transmembrane region.29
Pannarale described one family with an Arg135Trp mutation in rhodopsin with a very severe course. That family demonstrated non-recordable electroretinographic responses by the third decade of life but low, recordable wave forms for younger patients.17 Thus, changes associated with codon 135 may result in a more severe course.30 The appearance of white dots in association with rhodopsin and, specifically, Arg135Trp rhodopsin related RP, was described by Souied as an early finding in that form of RP.31 The term retinitis punctata albescens (RPA) was originally used to denote a specific form of RP.32 Although some have questioned in the past whether RPA was a specific disorder or simply a phenotype or stage of classic RP,33 recently, RPA has been recognised as a specific autosomal recessive retinal degeneration that results from mutations in the gene RLBP1.34–36 Admittedly, though, the white dots in RPA, although similar, are clinically different from those observed in our family.
In summary, we present a family with RP caused by an Arg135Leu mutation in rhodopsin. This family demonstrated a particularly severe course that was worse than families with cytoplasmic or intradiscal mutations in rhodopsin. From a cross sectional study of patients, we inferred the progression of disease for this family. In the first decade of life, affected patients will be symptomatic without clinical signs on fundus examination. By the second decade, patients develop extensive fine white dots at the level of the retinal pigment epithelium in the absence of extensive RPE atrophy or bone spicules. The white dots fade by the third decade of life leaving bone spicules with widespread RPE atrophy. This appearance steadily worsens with increasing age and is associated with progressive decrease in visual field area. Patients uniformly developed cataracts by the second decade of life and appeared to respond favourably to cataract extraction. However, macular atrophy, as observed in our patients, is not uncommon in patients with changes at codon 135 of rhodopsin.7,17,30 Central acuity appeared to be closely related to the degree of macular atrophy.17
Description of disease natural history is becoming increasingly important with the future of hereditary retinal degenerations directed towards intervention. This family demonstrates how a specific phenotype may be present only for a brief period in a patient’s life. While this is not the first paper to describe the association of a white dot phenotype with rhodopsin, we are the first to clearly document that this clinical appearance is largely dependent on the age of the subject. We also further demonstrated the importance of age and genotype for the modelling of disease course as previously described.37 The R2 value predicts that up to 40.7% of disease variation may be predicted on the basis of patient age and specific genotype. While other factors will influence disease course, this value calls attention to the importance of these two variables. The severity of this particular form of retinitis pigmentosa and the careful understanding of its natural history will be important considerations in the inclusion of patients with this condition in future clinical trials.
Acknowledgments
Supported in part by a career development award from the Foundation Fighting Blindness (KTO) and unrestricted grants from the Foundation Fighting Blindness (RGW and EMS), Research to Prevent Blindness (RGW and EMS). The authors thank the assistance of Gary Weaver, MD, Donald Bowden, PhD, and Joan Winnicki from Wake Forest University in identifying this family for further studies. The authors would also like to thank Cassandra Janowski, Andrea Billingslea, and Azadeh Rohanian Perry for their assistance in manuscript preparation and patient care.
Abbreviations
ADRP, autosomal dominant retinitis pigmentosa
ARMS, amplification refractory mutation system
GVF, Goldmann visual fields
mfERG, multifocal electroretinography
RP, retinitis pigmentosa
RPA, retinitis punctata albescens
RPE, retinal pigment epithelium
REFERENCES
- 1.Sohocki MM, Daiger SP, Bowne SJ, et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum Mutat 2001;17:42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bunker CH, Berson EL, Bromley WC, et al. Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 1984;97:357–65. [DOI] [PubMed] [Google Scholar]
- 3.Dickinson P , Mulhall L. A survey of hereditary aspects of pigmentary retinal dystrophies. Aust N Z J Ophthalmol 1989;17:247–56. [DOI] [PubMed] [Google Scholar]
- 4.Sung C-H, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA 1991;88:6481–6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sandberg MA, Weigel-DiFranco C, Dryja TP, et al. Clinical expression correlates with location of rhodopsin mutation in dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 1995;36:1934–42. [PubMed] [Google Scholar]
- 6.Sung C-H, Schneider BG, Agarwal N, et al. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA 1991;88:8840–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobson SG, Kemp CM, Sung C-H, et al. Retinal function and rhodopsin levels in autosomal dominant retinitis pigmentosa with rhodopsin mutations. Am J Ophthalmol 1991;112:256–71. [DOI] [PubMed] [Google Scholar]
- 8.Marmor MF, Zrenner E, for the Standardization Committee of the International Society for Clinical Electrophysiology of Vision (ISCEV. Standard for clinical electroretinography (1994 update). Doc Ophthalmol 1995;89:199–210. [DOI] [PubMed] [Google Scholar]
- 9.Marmor MF, Hood DC, Keating D, et al. Guidelines for basic multifocal electroretinography (mfERG). Doc Ophthalmol 2003;106:105–15. [DOI] [PubMed] [Google Scholar]
- 10.Weleber RG, Tobler WR. Computerized quantitative analysis of kinetic visual fields. Am J Ophthalmol 1986;101:461–8. [DOI] [PubMed] [Google Scholar]
- 11.Oh KT, Weleber RG, Lotery A, et al. Description of a new mutation in rhodopsin, Pro23Ala, and comparison with electroretingraphic and clinical characteristics of the Pro23His mutation. Arch Ophthalmol 2000;118:1269–76. [DOI] [PubMed] [Google Scholar]
- 12.Newton CR, Graham A, Heptinstall LE, et al. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS). Nucleic Acids Res 1989;17:2503–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bassam BJ, Caetano-Anolles G, Gresshoff PM. Fast and sensitive silver staining of DNA in ployacrylamide gels. Anal Biochem 1991;196:80–3. [DOI] [PubMed] [Google Scholar]
- 14.Buffone GJ, Darlington GJ. Isolation of DNA from biological specimens without extraction with phenol. Clin Chem 1985;31:164–5. [PubMed] [Google Scholar]
- 15.Andréasson S , Ehinger B, Abrahamson M, et al. A six-generation family with autosomal dominant retinitis pigmentosa and a rhodopsin gene mutation (arginine-135-leucine). Ophthalmic Paediatrics and Genetics 1992;13:145–153. [DOI] [PubMed] [Google Scholar]
- 16.Dryja TP. Doyne Lecture. Rhodopsin and autosomal dominant retinitis pigmentosa. Eye 1992;6:1–10. [DOI] [PubMed] [Google Scholar]
- 17.Pannarale MR, Grammatico B, Iannaccone A, et al. Autosomal dominant retinitis pigmentosa associated with an Arg-135-Trp point mutation of the rhodopsin gene: clinical features and longitudinal observations. Ophthalmology 1996;103:1443–52. [DOI] [PubMed] [Google Scholar]
- 18.Fishman GA, Stone EM, Sheffield VC, et al. Ocular findings associated with rhodopsin gene codon 17 and codon 182 transition mutations in dominant retinitis pigmentosa. Arch Ophthalmol 1992;110:54–62. [DOI] [PubMed] [Google Scholar]
- 19.Fishman GA, Stone EM, Gilbert LD, et al. Ocular findings associated with a rhodopsin gene codon 106 mutation: glycine-to-arginine change in autosomal dominant retinitis pigmentosa. Arch Ophthalmol 1992;110:646–53. [DOI] [PubMed] [Google Scholar]
- 20.Fishman GA, Stone EM, Gilbert LD, et al. Ocular findings associated with a rhodopsin gene codon 58 transversion mutation in autosomal dominant retinitis pigmentosa. Arch Ophthalmol 1991;109:1387–93. [DOI] [PubMed] [Google Scholar]
- 21.Heckenlively JR, Rodriguez JA, Daiger SP. Autosomal dominant sectoral retinitis pigmentosa: two families with transversion mutation in codon 23 of rhodopsin. Arch Ophthalmol 1991;109:84–91. [DOI] [PubMed] [Google Scholar]
- 22.Moore AT, Fitzke FW, Kemp CM, et al. Abnormal dark adaptation kinetics in autosomal dominant sector retinitis pigmentosa due to rod opsin mutation. Br J Ophthalmol 1992;76:465–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kranich H , Bartowski S, Denton MJ, et al. Autosomal dominant “sector” retinitis pigmentosa due to a point mutation predicting an Asn-15-Ser substitution of rhodopsin. Hum Mol Genet 1993;2:813–14. [DOI] [PubMed] [Google Scholar]
- 24.Dryja TP, Berson EL, Rao V, et al. Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat Genet 1993;4:280–3. [DOI] [PubMed] [Google Scholar]
- 25.Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol 2000;130:547–63. [DOI] [PubMed] [Google Scholar]
- 26.Sieving PA, Richards JE, Naarendorf F, et al. Dark-light: model for nightblindness from the human rhodopsin Gly-90 ∅ Asp mutation. Proc Natl Acad Sci USA 1995;92:880–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sieving PA, Fowler ML, Bush RA, et al. Constitutive "light" adaptation in rods from G90D rhodopsin: a mechanism for human congenital nightblindness without rod cell loss. J Neurosci 2001;21:5449–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berson EL, Rosner B, Weigel-DiFranco C, et al. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Invest Ophthalmol Vis Sci 2002;43:3027–36. [PubMed] [Google Scholar]
- 29.Berson EL, Rosner B, Sandberg MA, et al. Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine. Am J Ophthalmol 1991;111:614–23. [DOI] [PubMed] [Google Scholar]
- 30.Ponjavic V , Abrahamson M, Andreasson S, et al. Autosomal dominant retinitis pigmentosa with a rhodopsin mutation (Arg-135-Trp). Acta Ophthalmol Scand 1997;75:218–23. [DOI] [PubMed] [Google Scholar]
- 31.Souied E , Soubrane G, Benlian P, et al. Retinitis punctata albescens associated with the Arg135Try mutation in the rhodopsin gene. Am J Ophthalmol 1996;121:19–25. [DOI] [PubMed] [Google Scholar]
- 32.Lauber H . Die sogenannte Retinitis punctata albescens. Klin Monatsbl Augenheilkd 1910;48:133–48. [Google Scholar]
- 33.Deutman AF. Rod-cone dystrophy: primary, hereditary, pigmentary retinopathy, retinitis pigmentosa. In: Krill AE, ed. Krill’s hereditary retinal and choroidal diseases. New York: Harper & Row, 1977:479–576.
- 34.Morimura H , Berson EL, Dryja TP. Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Invest Ophthalmol Vis Sci 1999;40:1000–4. [PubMed] [Google Scholar]
- 35.Burstedt MS, Sandgren O, Holmgren G, et al. Bothnia dystrophy caused by mutations in the cellular retinaldehyde-binding protein gene (RLBP1) on chromosome 15q26. Invest Ophthalmol Vis Sci 1999;40:995–1000. [PubMed] [Google Scholar]
- 36.Eichers ER, Green JS, Stockton DW, et al. Newfoundland rod-cone dystrophy, an early-onset retinal dystrophy, is caused by splice-junction mutations in RLBP1. Am J Hum Genet 2002;70:955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oh KT, Longmuir R, Oh DM, et al. Comparison of the clinical expression of retinitis pigmentosa associated with rhodopsin mutations at codon 347 and codon 23. Am J Ophthalmol 2003;136:306–13. [DOI] [PubMed] [Google Scholar]