

Abstract
Background: The authors recently identified three large genetically unrelated families with an identical 17 base pair duplication mutation in exon 4 of the PITX3 gene. Here, they report the detailed clinical phenotype.
Methods: Affected and unaffected individuals in the three families with autosomal dominant posterior polar cataract underwent full clinical examination and donated blood samples for DNA extraction and molecular genetic studies.
Results: In all three families, an identical 17 base pair duplication mutation in PITX3 was identified which co-segregated with disease status in the family. All affected individuals had bilateral progressive posterior polar cataracts. In one family, posterior polar cataract was the only clinical abnormality but in the other two families, one of 10 affected individuals and four of 11 affected individuals also had anterior segment mesenchymal dysgenesis (ASMD).
Conclusion: Mutations in the PITX3 gene in humans result in posterior polar cataract and variable ASMD. The gene encodes a transcription factor which has a key role in lens and anterior segment development. The mechanism by which the mutant protein gives rise to such a regional pattern of lens opacity remains to be elucidated.
Keywords: congenital cataract, genetics, anterior segment dysgenesis, transcription factor
Cataracts are the commonest cause of blindness worldwide.1 They are broadly divided into adult onset and early onset (congenital or juvenile). Congenital cataract is responsible for approximately one tenth of worldwide childhood blindness.2 The incidence of congenital cataract is between 2.2 and 2.49 per 10 000 live births.3,4 About one third of isolated congenital cataracts are familial,5 the most common mode of inheritance being autosomal dominant.4,5 Cataracts may be inherited as isolated abnormalities, as part of a more complex ocular developmental abnormality, or as part of a systemic syndrome. A number of genes and loci have been implicated in isolated inherited cataract.6 The genes encode four major classes of protein: crystallins, transmembrane proteins, cytoskeletal proteins and transcription factors.
Transcription factors play an important role in the embryological development of the lens including the interaction between the embryonic surface ectoderm and the budding optic vesicle. This interaction is critical for normal lens induction.7 Mutations in several transcription factor genes notably PITX3,8PAX6,9FOXE3,10EYA1,11 and MAF12,13 have been implicated in congenital cataract and anterior segment mesenchymal dysgenesis (ASMD). The occurrence of both cataract and ASMD highlights the role of the lens in anterior segment development. The lens and the anterior segment are linked developmentally with a commonality of origin in surface ectoderm. In addition, there is a necessity for the separation of the corneal and lens surfaces for anterior chamber formation and a requirement for signalling from the lens for anterior segment organisation.14
In this paper, we present three genetically unrelated large families with autosomal dominant posterior polar cataract. We recently published the molecular genetic results.8 In all three families the causative mutation is a 17 base pair duplication mutation in exon 4 of PITX3. Here, we present a detailed description of the clinical phenotype associated with this mutation.
PATIENTS AND METHODS
The families in this study were identified from the genetic clinic database at Moorfields Eye Hospital. This provided details of the probands, from whom the pedigrees were constructed. Moorfields ethics committee approval was obtained for the studies and all individuals taking part in the study gave written informed consent.
Both affected and unaffected individuals underwent full clinical examination. This included comprehensive ophthalmic examination, with careful slit lamp examination and anterior segment photography when appropriate. Peripheral blood samples were collected from which DNA was extracted for subsequent molecular genetic analysis. The molecular genetic methodology has been described previously.8
The pedigrees are shown in figure 1. All three families are of English descent and autosomal dominant inheritance is supported by the presence of affected male and female individuals in each generation and male to male transmission. The following number of individuals were examined from each family: eight affected and 11 unaffected from family A, 10 affected and nine unaffected from family B, 14 affected and 18 unaffected from family C. The age ranges of the individuals examined were: 8–70 years (family A), 8–70 (family B), 7–80 (family C). Overall, in the three families, 30 male and 42 female individuals were examined.
Figure 1.
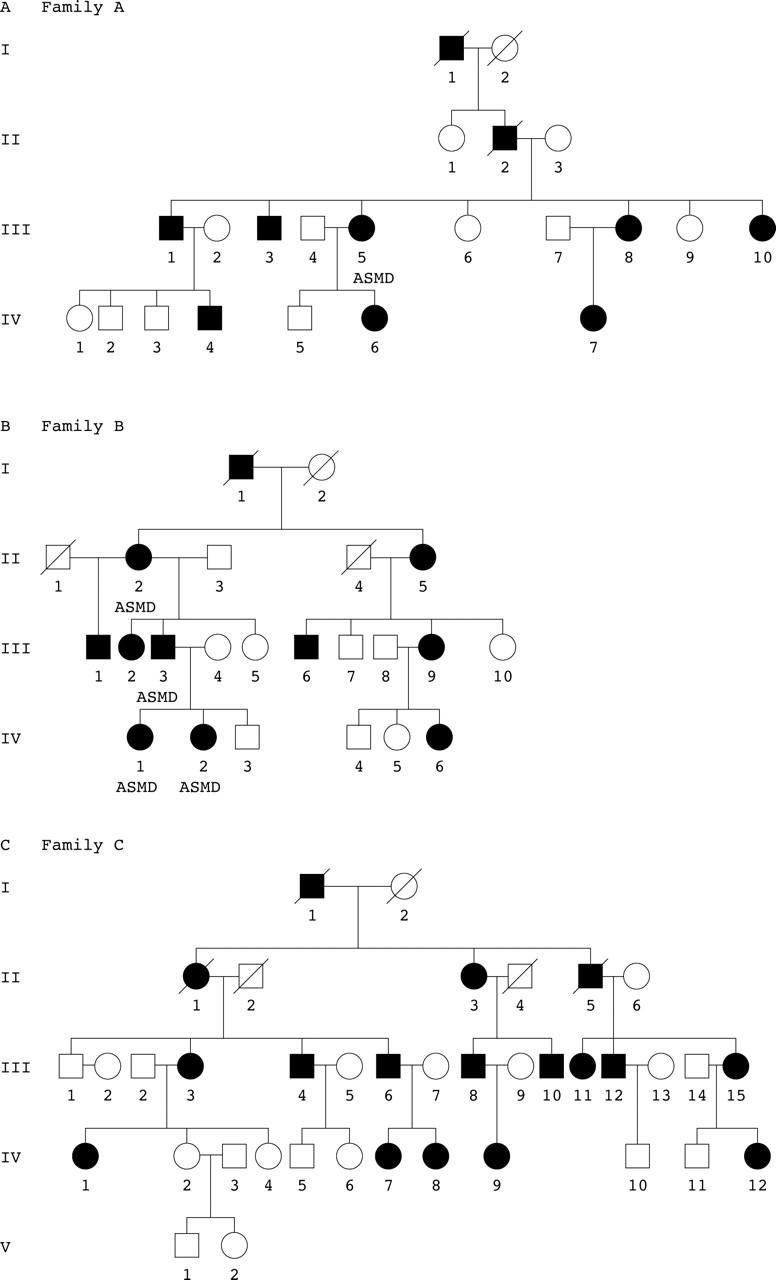
Pedigrees: families (A), (B), and (C). Square symbols denote males; circles denote females; affected individuals are denoted by black symbols.
RESULTS
The molecular genetic studies, reported elsewhere,8 showed that all affected individuals in each of the three families had an identical mutation in the PITX3 gene. Sequence analysis of this gene revealed a 17 base pair duplication mutation in exon 4 which co-segregated with the disease phenotype in the families. The mutation results in a frameshift in codon 220 and leads to the production of an aberrant protein consisting of 94 additional residues. Haplotyping confirmed that the three families are unrelated.
All affected individuals from the three families had the same cataract phenotype. Posterior polar cataract was fully penetrant and exhibited some variable expressivity. The typical natural history was to develop gradually progressive bilateral posterior polar cataracts (fig 2A and B), which became clinically significant within the first decade of life and visually significant within the first two decades, necessitating cataract extraction and lens implantation.
Figure 2.

(A) Direct illumination and (B) retroillumination slit lamp photographs of posterior polar cataract in individual IV:7 from family A. (C) Right and (D) left eyes of patient III:5 from family A at the end of repeat left penetrating keratoplasty. (E) Microscope photograph of individual IV:1 from family B, showing central corneal opacity with associated iridocorneal adhesions. (F) Slit lamp photograph of individual II:2 from family B showing complex anterior segment dysgenesis with corneal opacity and dense cataract.
A few individuals affected with cataract also exhibited features of ASMD. In family A, one of 10 affected individuals had ASMD (subject III:5). This subject had, in addition to cataracts, congenital glaucoma, posterior embryotoxon, atrophic irides, and microcornea. She underwent a left penetrating keratoplasty aged 28 and repeat penetrating keratoplasty aged 41 for endothelial decompensation in the previous graft (fig 2C and D). In family B, four affected individuals had ASMD. There was considerable variability in the ASMD with features ranging from superior sclerocornea to central corneal opacity with iridocorneal adhesions (fig 2E and F). The visual acuities of the individuals with cataract alone ranged from 6/5 to light perception (in the unoperated eyes) and from 6/5 to light perception (in the operated eyes). The visual acuities of the subjects with cataract and ASMD ranged from hand movements to light perception (in the unoperated eyes) and from 6/6 to 6/60 (in the operated eyes).
DISCUSSION
We have described three genetically unrelated families with autosomal dominant progressive posterior polar cataract. They all have an identical 17 base pair (17 bp) duplication mutation in the PITX3 gene. Cataract causing mutations in PITX3 have only been previously reported in two other families.15 One was a large family with an identical 17 bp duplication mutation causing cortical cataracts and ASMD in all of the affected individuals. The second was a small family with total cataract in which both affected individuals developed glaucoma at a young age. In this paper, we report a large family in which all the affected individuals with the 17 bp duplication mutation have posterior polar cataract without associated ASMD. Posterior polar cataract has not previously been reported in association with mutations in PITX3. Autosomal dominant posterior polar cataract is genetically heterogeneous. Two loci on 1p36,16 and 20p1217 and mutations in one gene, CRYAB on 11q21,18 have been reported in association with this phenotype. We have demonstrated that mutations in PITX3, on 10q25, can give rise to an isolated posterior polar cataract phenotype. The mechanism by which mutations in PITX3 give rise to such a localised form of lens opacity remains unknown.
Mutations in a number of genes encoding transcription factors including PITX3,8PAX6,9FOXE3,10 EYA1,11 and MAF12,13 have been implicated in ASMD and cataract. Most mutations are associated with ASMD and cataract in all affected individuals within the family although there is variability in the phenotype. A single family with a mutation in MAF has been reported in which some affected members of the family had both ASMD and cataract, while others had cataract alone.12,13 Another transcription factor gene, HSF4,19 has been reported to cause cataract alone in all affected individuals within a large family.
PITX3 is a member of the PITX gene family and encodes a paired-like class of homeobox transcription factors. Both PITX2 and PITX3 genes are involved in eye development and are expressed in most developing ocular tissues, including the cornea, lens, and retina.20 Mutations in PITX2 have been reported in Rieger’s syndrome.21 The 17 bp duplication mutation in exon 4 of PITX3, reported here, results in a frameshift in codon 220 and leads to the production of an aberrant protein consisting of 94 additional residues. The mutation does not affect the homeodomain and the mechanism by which the aberrant protein leads to cataract and ASMD is unclear. A better understanding of the disease mechanism will be gained by future functional studies.
In two of the three families reported here, affected members showed a wide variation in phenotype. Some affected individuals had cataract alone whereas others had ASMD of variable severity. The anterior segment developmental abnormalities varied from peripheral sclerocornea to complex disorders with changes similar to Peter’s anomaly (central corneal opacity with iridocorneal adhesions). This variability between individuals carrying the same causative mutation may result from a number of factors including the effects of other modifier genes, intrauterine environmental factors, or stochastic developmental events.22
Acknowledgments
We thank the family members for taking part in these studies. This work is supported by The Wellcome Trust, project grant number: 063969/Z/01.
SSB thanks The Special Trustees of Moorfields Eye Hospital for their general support.
Abbreviations
ASMD, anterior segment mesenchymal dysgenesis
Competing interests: The authors have no competing interests.
Ethical approval: Ethics committee approval was obtained from The Moorfields Eye Hospital research ethics committee. The project number is MOOA183.
REFERENCES
- 1.World Health Organization. The world health report 1998: life in the 21st century: a vision for all. Geneva: WHO, 1998.
- 2.Gilbert CE, Canovas R, Hagan M, et al. Causes of childhood blindness: results from West Africa, south India and Chile. Eye 1993;7:184–8. [DOI] [PubMed] [Google Scholar]
- 3.Rahi JS, Dezateaux C. Measuring and interpreting the incidence of congenital ocular anomalies: lessons from a national study of congenital cataract in the UK. Invest Ophthalmol Vis Sci 2001;42:1444–8. [PubMed] [Google Scholar]
- 4.Wirth MG, Russell-Eggitt IM, Craig JE, et al. Aetiology of congenital and paediatric cataract in an Australian population. Br J Ophthalmol 2002;86:782–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rahi JS, Dezateaux C. Congenital and infantile cataract in the United Kingdom: underlying or associated factors. British Congenital Cataract Interest Group. Invest Ophthalmol Vis Sci 2000;41:2108–14. [PubMed] [Google Scholar]
- 6.Reddy MA, Francis PJ, Berry V, et al. Molecular genetic basis of inherited cataract and associated phenotypes. Surv Ophthalmol 2004;49:300–15. [DOI] [PubMed] [Google Scholar]
- 7.Ogino H, Yasuda K. Sequential activation of transcription factors in lens induction. Dev Growth Differ 2000;42:437–48. [DOI] [PubMed] [Google Scholar]
- 8.Berry V, Yang Z, Addison PKF, et al. Recurrent 17bp duplication in PITX3 is primarily associated with posterior polar cataract (CPP4). J Med Genet 2004;41:e109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hanson I, Churchill A, Love J, et al. Missense mutations in the most ancient residues of the PAX6 paired domain underlie a spectrum of human congenital eye malformations. Hum Mol Genet 1999;8:165–72. [DOI] [PubMed] [Google Scholar]
- 10.Semina EV, Brownell I, Mintz-Hittner HA, et al. Mutations on the human forkhead transcription factor FOXE3 associated with anterior segment ocular dysgenesis and cataracts. Hum Mol Genet 2001;10:231–6. [DOI] [PubMed] [Google Scholar]
- 11.Azuma N, Hirakiyama A, Inoue T, et al. Mutations of a human homologue of the Drosophila eyes absent gene (EYA1) detected in patients with congenital cataracts and ocular anterior segment anomalies. Hum Mol Genet 2000;9:363–6. [DOI] [PubMed] [Google Scholar]
- 12.Jamieson RV, Perveen R, Kerr B, et al. Domain disruption and mutation of the bZIP transcription factor, MAF, associated with cataract, ocular anterior segment dysgenesis and coloboma. Hum Mol Genet 2002;11:33–42. [DOI] [PubMed] [Google Scholar]
- 13.Jamieson RV, Munier F, Balmer A, et al. Pulverulent cataract with variably associated microcornea and iris coloboma in a MAF mutation family. Br J Ophthalmol 2003;87:411–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beebe DC, Coats JM. The lens organizes the anterior segment: specification of neural crest cell differentiation in the avian eye. Dev Biol 2000;220:424–31. [DOI] [PubMed] [Google Scholar]
- 15.Semina EV, Ferrell RE, Mintz-Hittner HA, et al. A novel homeobox gene PITX3 is mutated in families with autosomal-dominant cataracts and ASMD. Nat Genet 1998;19:167–70. [DOI] [PubMed] [Google Scholar]
- 16.Ionides AC, Berry V, Mackay DS, et al. A locus for autosomal dominant posterior polar cataract on chromosome 1p. Hum Mol Genet 1997;6:47–51. [DOI] [PubMed] [Google Scholar]
- 17.Yamada K, Tomita HA, Kanazawa S, et al. Genetically distinct autosomal dominant posterior polar cataract in a four-generation Japanese family. Am J Ophthalmol 2000;129:159–65. [DOI] [PubMed] [Google Scholar]
- 18.Berry V, Francis P, Reddy MA, et al. Alpha-B crystalline gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet 2001;69:1141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bu L, Jin Y, Shi Y, et al. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet 2002;31:276–8. [DOI] [PubMed] [Google Scholar]
- 20.Gage PJ, Suh H, Camper SA. The bicoid-related Pitx gene family in development. Mamm Genome 1999;10:197–200. [DOI] [PubMed] [Google Scholar]
- 21.Semina EV, Reiter R, Leysens NJ, et al. Cloning and characterization of a novel bicoid-related homeobox transcription factor gene, RIEG, involved in Rieger syndrome. Nat Genet 1996;14:392–9. [DOI] [PubMed] [Google Scholar]
- 22.Smith RS, Zabaleta A, Kume T, et al. Haploinsufficiency of the transcription factors FOXC1 and FOXC2 results in aberrant ocular development. Hum Mol Genet 2000;9:1021–32. [DOI] [PubMed] [Google Scholar]