

Abstract
Aim: To characterise the phenotype of an autosomal dominant cone-rod dystrophy (CORD7) associated with the Arg844His mutation in RIM1.
Methods: Eight members of a four generation, non-consanguineous British family were examined clinically and underwent electrophysiological testing, automated dark adapted perimetry, dark adaptometry, colour vision assessment, colour fundus photography, fundus fluorescein angiography (FFA), and fundus autofluorescence (AF) imaging.
Results: The majority of affected individuals described a progressive deterioration of central vision, night vision, and peripheral visual field usually between the third and fourth decades. The visual acuity ranged from 6/6 to 3/60. Colour vision testing showed mild to moderate dyschromatopsia in the majority of individuals. Fundus changes comprised a range of macular appearances varying from mild retinal pigment epithelial (RPE) disturbance to extensive atrophy and pigmentation. In some individuals retinal vessels were attenuated and in two subjects peripheral areas of retinal atrophy were present. An absent or severely reduced PERG was detected in all subjects, indicative of marked macular dysfunction. Full field ERG showed abnormal rod and cone responses. AF imaging revealed decreased macular AF centrally surrounded by a ring of increased AF in the majority of individuals. “Bull’s eye” lesions were present in two individuals, comprising of a ring of decreased perifoveal AF bordered peripherally and centrally by increased AF. Photopic sensitivity testing demonstrated elevated central visual field thresholds with additional superior greater than inferior peripheral field loss. There were rod and cone sensitivity reductions in the central and peripheral visual fields, with the inferior retina being more affected than the superior.
Conclusions: The detailed phenotype is described of the autosomal dominant cone-rod dystrophy, CORD7, which is associated with a point mutation in RIM1, a gene encoding a photoreceptor synaptic protein. The pattern of disease progression and long term visual outcome facilitates improved genetic counselling and advice on prognosis. Such phenotypic data will be invaluable in the event of future therapy.
Keywords: autosomal dominant cone-rod dystrophy
The cone and cone-rod dystrophies (CORD) are a clinically heterogeneous group of retinal disorders characterised by reduced central vision and photophobia. Pure cone dystrophies are uncommon and in most instances there is eventual rod involvement leading to poor night vision. The age of onset is variable, but many patients present in the first two decades of life.
CORDs are also genetically heterogeneous; families demonstrating autosomal dominant, autosomal recessive and X linked recessive inheritance have been reported, and a number of causative genes and chromosomal loci have now been identified (table 1). When an inheritance pattern can be reliably established, it is most commonly autosomal dominant (ad).21,22 Currently six genes have been shown to be associated with adCORD; CRX,2,3GUCY2D,5,6RIM1,7Peripherin/RDS,13GUCA1A,18 and AIPL1,20 with more remaining to be discovered. Mutations in ABCA4 are believed to be the commonest cause of autosomal recessive CORD.14,15
Table 1.
Cone-rod dystrophies (CORDs) with identified genes and known chromosomal loci
| CORD; OMIM number | Mode of inheritance | Chromosome locus | Mutated gene | Reference |
| CORD1; 600624 | Autosomal recessive | 18q21.1–q21.3 | Not identified | 1 |
| CORD2; 120970 | Autosomal dominant | 19q13.1–q13.2 | CRX | 2, 3 |
| CORD3; 604116 | Autosomal recessive | 1p21–p13 | ABCA4 | 4 |
| CORD5 and CORD6; 600977 and 601777 | Autosomal dominant | 17p13–p12 | GUCY2D | 5, 6 |
| CORD7; 603649 | Autosomal dominant | 6q14 | RIM1 | 7 |
| CORD8; 605549 | Autosomal recessive | 1q12–q24 | Not identified | 8 |
| CORD | Autosomal recessive | 8p11 | Not identified | 9 |
| CORD; 304020 | X linked | Xp21.1–p11.3 (COD1) | RPGR | 10, 11 |
| CORD | X linked | Xp11.4–q13.1 (COD4) | Not identified | 12 |
| CORD; 179605 | Autosomal dominant | 6p21.2-cen | Peripherin/RDS | 13 |
| CORD; 601691 | Autosomal recessive | 1p21–p13 | ABCA4 | 14, 15 |
| CORD9; 608194 | Autosomal recessive | 14q11 | RPGRIP1 | 16 |
| CORD; 600053 | Autosomal recessive | 2q11 | CNGA3 | 17 |
| CORD; 600364 | Autosomal dominant | 6p21.1 | GUCA1A | 18 |
| CORD; 604011 | – | 17q11.2 | UNC119 | 19 |
| CORD; 604392 | Autosomal dominant | 17p13.1 | AIPL1 | 20 |
The adCORD, CORD7, was originally mapped in a four generation, non-consanguineous British family to a 7 cM region of chromosome 6q14, flanked by the markers D6S430 and D6S1625.23 Recently a candidate gene screening approach has identified a mutation in the Rab3A interacting molecule (RIM1) gene in CORD7.7 The G to A point mutation results in an Arg844His substitution in the highly conserved C2A domain of the RIM1 protein and was found to segregate with disease. RIM1 is expressed in brain and retinal photoreceptors where it is localised to the pre-synaptic ribbons in ribbon synapses, with the protein product believed to play an important part in synaptic transmission and plasticity.24–26
A detailed study of the CORD7 phenotype has been performed in order to improve genetic counselling and advice on prognosis, and also with the hope of assisting our understanding of the effects of the RIM1 mutation on retinal function.
PATIENTS AND METHODS
Patients
A four generation, non-consanguineous British family with an autosomal dominant cone-rod dystrophy (CORD7, OMIM 603649) was ascertained (fig 1). After informed consent, a detailed medical and ophthalmic history was obtained and a full ophthalmological examination performed. Subjects also underwent electrophysiological testing, automated photopic and dark adapted perimetry, dark adaptometry, colour vision assessment, colour fundus photography, and fundus autofluorescence imaging. Individuals were diagnosed as affected on the basis of the presence of retinal abnormality and in most cases associated decreased visual acuity. The protocol of the study was approved by the local ethics committee.
Figure 1.

Four generation pedigree of a British family with autosomal dominant cone-rod dystrophy (CORD7).
Methods
Fundus autofluorescence imaging was undertaken using the confocal scanning laser ophthalmoscope (cSLO) (Zeiss Prototype; Carl Zeiss Inc, Oberkochen, Germany). Electrophysiological assessment included an electro-oculogram (EOG), a full field electroretinogram (ERG), and pattern ERG (PERG), incorporating the protocols recommended by the International Society for Clinical Electrophysiology of Vision.27–29 Colour vision testing was performed using the Hardy, Rand, Rittler (HRR) plates (American Optical Company, NY, USA).
Five affected subjects underwent detailed perimetry and dark adaptation. Static threshold perimetry in the dark and light adapted states was performed using a Humphrey field analyser (Allergan Humphrey, Hertford, UK). For dark adapted visual fields, the pupil was dilated with 2.5% (w/v) phenylephrine hydrochloride and 1% (w/v) tropicamide, and the patient was dark adapted for 45 minutes. The Humphrey field analyser was modified for use in dark adapted conditions.30–32 An infrared source illuminated the bowl, and an infrared monitor (Phillips, Eindhoven, Netherlands) was used to detect eye movements. Fields were recorded using the 30–2, peripheral 30/60–2, and macular programs. The target size corresponded to Goldmann size V for peripheral testing and to Goldmann size III for macular programs. Each was performed with a red (dominant wavelength, 650 nm) and then blue (dominant wavelength, 450 nm) filter in the stimulus beam.
For dark adaptometry, two test locations were chosen at 3° and 9°. The Humphrey field analyser used was controlled by a custom computerised program (PS/2 model 50; International Business Machines, Armonk, NY, USA).32,33 Fully dark adapted rod thresholds were measured before exposure to the adapting light at the two coordinates with the blue filter in the stimulus beam.
RESULTS
The autosomal dominantly inherited CORD7 was present in a four generation British family as shown in figure 1. Patients II:3, II:8, and IV:1, were asymptomatic, had normal clinical examination, and did not carry the RIM1 mutation; and were therefore designated as unaffected. All the affected individuals studied in detail carried the RIM1 mutation. The clinical findings are summarised in table 2, with representative fundus and autofluorescence (AF) images illustrated in figures 2 and 5.
Table 2.
Summary of clinical findings
| Patient | Sex | Age | Visual acuityRE-LE | Age of onset | Fundus | AF imaging | ERG | PERG | Colour vision |
| II:2 | F | 81 | 6/60–6/60 | 20 | Bilateral symmetrical macular atrophy and attenuated retinal vessels (fig 2E) | Bilateral decreased AF corresponding to atrophy seen on ophthalmoscopy with a surrounding ring of relative increased AF | Reduced cone and rod responses | Absent | Bilateral medium protan, deutan and tritan defects |
| II:5 | F | 68 | 3/36–3/60 | 33 | Bilateral macular atrophy; attenuated retinal vessels and peripheral areas of atrophy (fig 4) | Bilateral decreased AF corresponding to atrophy seen on ophthalmoscopy with a surrounding ring of relative increased AF | Markedly reduced cone and absent rod responses | Absent | Bilateral medium protan, deutan and tritan defects |
| II:7 | F | 75 | 6/9–6/9 | 41 | Bilateral “bull’s eye maculopathy” | Concentric rings of increased and decreased AF | Borderline reduced maximal responses | Reduced | Bilateral mild protan, deutan and tritan defects |
| III:1 | M | 52 | 6/36–6/36 | 30 | Bilateral macular RPE changes with areas of atrophy and pigmentation; attenuated retinal vessels with peripheral areas of atrophy (fig 4) | Bilateral decreased AF centrally with a surrounding ring of relative increased AF | Cone responses more markedly reduced than rod | Absent | Bilateral mild protan, deutan and tritan defects |
| III:2 | M | 48 | 6/9–6/9 | 42 | RE: RPE changes | RE: Concentric rings of increased and decreased AF | Borderline reduction | Absent | Bilateral mild protan, deutan and tritan defects |
| LE: Well defined area of macular atrophy (fig 2A) | LE : Decreased AF corresponding to atrophy seen on ophthalmoscopy with surrounding ring of relative increased AF (fig 2B) | ||||||||
| III:6 | F | 53 | 3/60–6/60 | 19 | Extensive RPE atrophy and areas of pigmentation RE > LE; and attenuated retinal vessels | Bilateral decreased AF centrally with a surrounding ring of relative increased AF | Cone responses more markedly reduced than rod | Absent | Bilateral medium protan, deutan and tritan defects |
| IV:2 | F | 18 | 6/6–6/6 | – | Bilateral mild macular RPE changes | Bilateral perifoveal rings of relative increased AF (fig 5) | Borderline reduction | Reduced | N |
| IV:3 | M | 31 | 6/18–3/60 | 14 | Extensive RPE atrophy and areas of pigmentation LE > RE (fig 2C) | Bilateral decreased AF centrally with a surrounding ring of relative increased AF (fig 2D) | Markedly reduced cone and rod responses | Absent | Bilateral mild protan, deutan and tritan defects |
BEM, bull’s eye maculopathy; IV:2 was asymptomatic; ERG, electro-retinography; PERG, pattern ERG.
Figure 2.
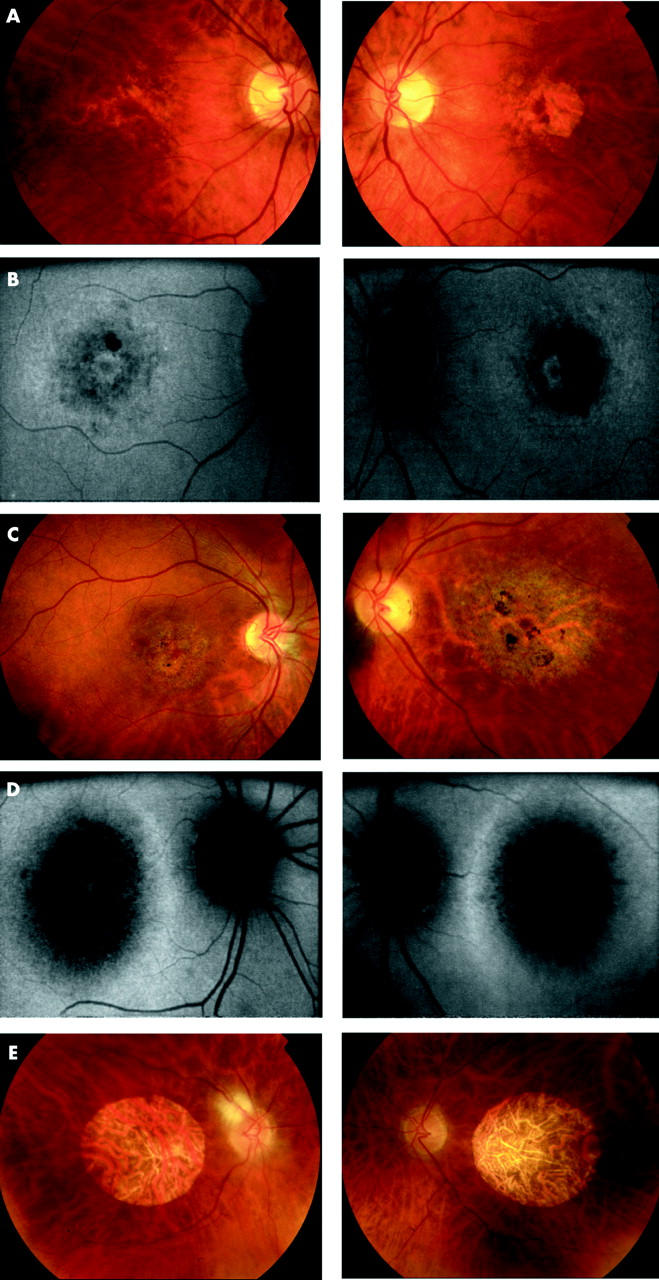
(A) Patient III:2 (48 years old) Fundus photographs showing bilateral macular RPE disturbance with a well defined area of atrophy at the left macula. (B) Patient III:2 (48 years old) Fundus autofluorescence imaging. RE: Concentric rings of increased and decreased AF in a BEM-like pattern. LE: Decreased AF corresponding to atrophy seen on ophthalmoscopy with a surrounding ring of relative increased AF. (C) Patient IV:3 (31 years old). Fundus photography showing extensive macular RPE atrophy and areas of pigmentation, worse in the left than right eye. (D) Patient IV:3 (31 years old). Fundus autofluorescence imaging showing bilateral decreased AF centrally with a surrounding ring of relative increased AF. (E) Patient II:2 (81 years old). Fundus photographs showing bilateral symmetrical well circumscribed macular atrophy and attenuated retinal vessels.
Figure 5.

Patient IV:2 (18 years old). Fundus autofluorescence imaging showing bilateral perifoveal rings of relative increased AF.
The age of onset of symptoms was variable and ranged from the second to the fifth decade of life. Most individuals described a progressive deterioration in central vision, night vision, and peripheral visual field constriction especially during the third and fourth decades. Affected subjects experienced mild photophobia and had no nystagmus. Mild to moderate generalised dyschromatopsia was detected in the majority of individuals. A range of macular appearances was seen, varying from mild retinal pigment epithelial (RPE) disturbance to extensive atrophy and pigmentation (table 2, fig 2). Subject II:5 had previously been noted to have a bull’s eye maculopathy (BEM) appearance and a fluorescein angiogram performed at that time (aged 42 years) was consistent with this (fig 3). On review her fundus appearance had progressed, with extensive macular atrophy now being present. Retinal vessels were attenuated in four individuals and peripheral areas of retinal atrophy were present in two subjects (fig 4).
Figure 3.

Fundus photographs and FFA of II:5 (42 years old), showing a BEM appearance.
Figure 4.

Fundus photographs of II:5 (left, 68 years old) and III:1 (right, 52 years old) showing areas of peripheral retinal atrophy.
AF imaging in the majority of individuals revealed decreased macular AF centrally surrounded by a ring of increased AF (figs 2 and 5). The decreased AF corresponded to the retinal atrophy seen ophthalmoscopically. Bull’s eye lesions were present in two individuals, consisting of a ring of decreased perifoveal autofluorescence bordered peripherally and centrally by increased autofluorescence (fig 2B).
Representative electrophysiological traces appear in figure 6. The EOG was normal in all affected subjects. The pattern ERG was abnormal in all affected individuals, in keeping with macular dysfunction, and the full field ERG showed abnormal cone and rod responses. The ERG was variably affected and in three mildly affected individuals, full field ERGs were only marginally abnormal. In most individuals cone amplitudes were reduced to a greater extent than rod. The 30 Hz cone flicker ERG implicit times were normal in all subjects except IV:3, in whom implicit time was delayed. Delayed recovery from the peak of the photopic b-wave, with no evidence of an i-wave, was seen in older individuals. No patients showed any suggestion of specific b-wave reduction; a negative ERG was not observed in any patient and in no patient was there any suggestion of a b:a ratio reduction. This can be seen in figure 6 where there is always parallel reduction in both a-waves and b-waves. The majority of patients showed similar degrees of ON- and OFF- response amplitude, with no consistent effect on either ON- or OFF- pathways. There is electrophysiological evidence of progressive deterioration in retinal function in subject IV:3 (fig 7).
Figure 6.

Electrophysiological traces from the patients examined with a normal (N) set of traces for comparison. All show marked central cone dysfunction with pattern ERG being undetectable in most, and markedly reduced in the remaining patients. Maximal response a-wave amplitude, a measure of rod photoreceptor function, is subnormal in all patients (normal >250 μV in young patients; >200 μV in older patients). There is marked variation in severity within generations. The 30 Hz flicker ERG is abnormal in most patients (normal >70 μV for younger patients; >50 μV in older patients), and is usually of normal implicit time, the only exceptions being IV:3 where there is delay, and III:6 where the values are borderline. The absence of the i-wave, clearly seen in the normal and younger patients after the descent of the photopic b-wave, is of uncertain significance. ON-OFF- ERGs and short wavelength cone ERGs (S-cone) generally parallel the conventional cone ERGs, without specific preservation of any cone subsystem. The OFF- response is marginally delayed in patients IV:3 and III:6.
Figure 7.

Electrophysiological data showing progressive deterioration of retinal function. ERG findings in patient IV:3 in 1999 (27 years old) and 2003 (31 years old) demonstrate clear deterioration in rod specific ERG b-wave amplitude, and reduction in maximal response a-wave amplitude reflecting increasing loss of rod photoreceptor function. There are also changes in full field cone derived ERGs. PERG was undetectable on both visits.
Perimetry and kinetics
Five affected individuals underwent detailed assessment. Photopic testing demonstrated decreased sensitivity, ranging from 2 to 3 log units, with the upper field being more severely affected (fig 8). Dark adapted perimetry revealed reduction in both rod and cone sensitivities, with well localised central loss, and peripheral loss being greater in the superior field than the inferior. Peripheral visual field sensitivity losses ranged from 6 to 10 dB (fig 9).
Figure 8.

Photopic 30° static threshold perimetry of IV:3 (31 years old) and III:2 (48 years old), showing central sensitivity reduction, with peripheral loss being more marked in the superior visual field. IV:3 is more severely affected than III:2.
Figure 9.
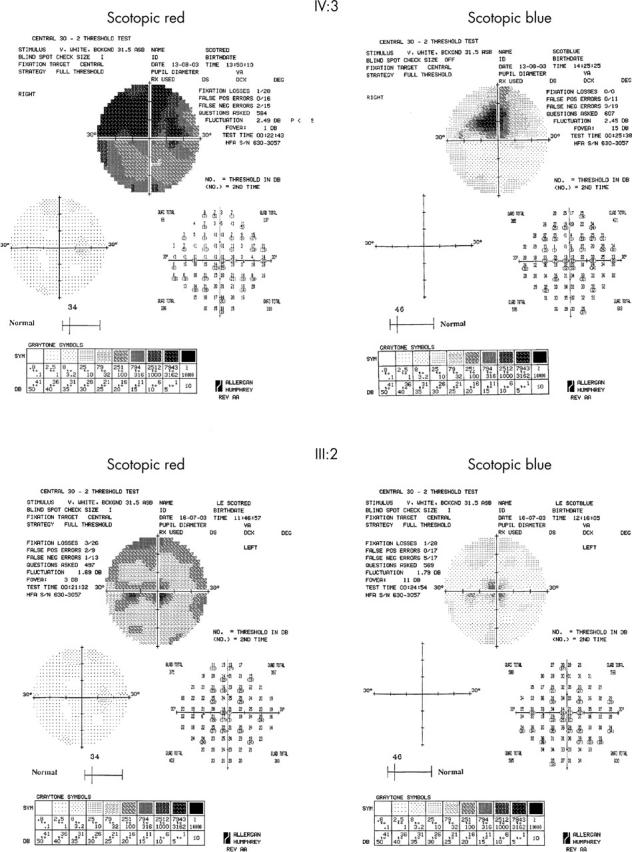
Scotopic red and blue 30° static threshold perimetry of IV:3 (31 years old) and III:2 (48 years old), showing central reduction of both cone and rod sensitivities, with peripheral loss being more marked in the inferior retina. IV:3 is more severely affected than III:2.
Dark adaptometry performed in mildly affected individuals showed rod final threshold elevation but no abnormality of kinetics. In more severely affected subjects both rod and cone threshold elevation was detected, and in one individual the rod-cone break was not detectable.
DISCUSSION
The detailed phenotype is described of the autosomal dominant cone-rod dystrophy, CORD7, associated with a point mutation in RIM1, a gene encoding a photoreceptor synaptic protein. The phenotype is characterised by progressive deterioration in central vision, night vision and peripheral visual field, ophthalmoscopic abnormalities generally confined to the macula, and electrophysiological and psychophysical evidence of widespread cone and rod abnormalities; findings consistent with a CORD. The age of onset is variable, ranging from the second to fifth decades of life, with a worse visual prognosis generally associated with an earlier age of onset (table 2). A more severe phenotype was not only observed in older individuals—for example, II:7 was a mildly affected 75 year old subject (grandmother), whereas IV:3 was severely affected at 31 years of age (her grandson). Both environmental factors and other genes modifying the effect of the RIM1 mutation may play a part in the observed variable severity. The funduscopic abnormalities were generally restricted to the macular region and ranged from mild RPE disturbance in the early stages to extensive chorioretinal atrophy and pigmentation.
Autofluorescence (AF) imaging allows the visualisation of the RPE by taking advantage of the fluorescent properties of lipofuscin.34,35 Affected subjects showed decreased AF corresponding to areas of atrophy seen ophthalmoscopically, which were encircled by a ring of increased AF. A perifoveal ring of increased AF was also detected in the youngest individual (18 years old), who had very mild RPE disturbance at the macula and was asymptomatic. The presence of a perifoveal ring may thus be an early indicator of affected status.
There is evidence of continuous degradation of autofluorescent material in the RPE.35 Progressive loss of lipofuscin occurs when there is reduced metabolic demand due to photoreceptor cell death, which results in areas of decreased AF in eyes with photoreceptor degeneration.35 It has been demonstrated histologically that the number of photoreceptors is reduced in the presence of increased quantities of lipofuscin in the RPE, in keeping with an accumulation of autofluorescent material before cell death.36,37 It is therefore likely that in our family the ring of increased AF may represent the front of advancing concentric photoreceptor cell loss. It has been established that there is reduced sensitivity across the similar appearing ring of increased AF that can occur in retinitis pigmentosa, and a similar mechanism proposed.38 This increase in lipofuscin may reflect either increased outer segment turnover or the inability of the RPE to process outer segment debris.
A BEM was present in two family members, better seen on AF imaging than on direct ophthalmoscopy, in keeping with previous reports.39,40 The pathogenesis of BEM is poorly understood. The characteristic appearance in which there is annular RPE atrophy and central sparing may correspond to the pattern of lipofuscin accumulation in the RPE, which in healthy individuals is highest at the posterior pole and shows a depression at the fovea.36 Bull’s eye maculopathy is a non-specific appearance that can occur in relation to a number of underlying disorders.21,39,41–44
The normal EOG suggests that there is no widespread dysfunction of the RPE, consistent with RIM1 being located at photoreceptor ribbon synapses. While the absent or markedly reduced PERG in all affected individuals demonstrates marked macular dysfunction. ERG testing revealed no evidence of impaired synaptic transmission per se; there being no suggestion of electronegative ERG waveforms. Maximal response a-waves were profoundly abnormal suggesting that the primary site of dysfunction lies at the level of the photoreceptors. The unusual finding of a normal 30 Hz flicker ERG implicit time suggests either that there is no significant abnormality of the cone phototransduction cascade, or that there is a normal recovery from photoactivation, such as has been proposed in the cone dystrophy consequent upon mutation in retinal guanylate cyclase activating protein (GCAP), in which a normal 30 Hz implicit time has also been observed.18 A second unusual finding in our study in older individuals was anomalous recovery from the peak of the photopic b-wave, with no evidence of an i-wave; the significance of which is unknown. In some subjects there was a similar reduction in both cone and rod full field ERG amplitudes. However, given the profound PERG abnormality in all individuals, we have preferred the term cone-rod dystrophy to reflect this marked central cone dysfunction.
The Arg844His missense mutation located in the highly conserved C2A domain of RIM1 was recently identified in this (CORD7) family.7 The protein RIM1 localises to the pre-synaptic active zone in conventional synapses and to pre-synaptic ribbons of ribbon synapses where it has a critical role in the tethering of synaptic vesicles,24,45,46 although a post-docking role in regulating vesicle priming has also been suggested.47,48 It is a large multidomain protein with different regions responsible for the different interactions that it undertakes. Key domains are the N-terminal Rab3A-GTP binding site responsible for vesicle binding via Rab3A-GTP and the two C-terminal C2A domains, that interact with other proteins in the synaptic complex, especially synaptotagmin. The effect of the Arg844His mutation in the C2A domain may be to alter the affinity of RIM1 for either the α1D subunit of L-type Ca2+ channels or synaptotagmin (synaptic vesicle associated Ca2+ sensor) and thereby the rate of synaptic vesicle docking and fusion in response to a Ca2+ signal.7 This may be expected to interfere with transmission at the photoreceptor synapse but electrophysiological testing of patients with RIM1 mutations showed no evidence of post-phototransduction dysfunction, even in subjects with early disease. It thus remains likely that mutant RIM1 adversely affects function at the photoreceptor side of the synaptic complex, although abnormality of synaptic transmission itself has not been detected with current techniques.
This is the second example of a mutation in a protein with a potential role in synaptic function causing a CORD phenotype; the first being a premature termination mutation in UNC119 (HRG4), a photoreceptor synaptic protein that is also highly enriched in ribbon synapses.19 It is also of interest that in a histopathological study of a patient with cone-rod dystrophy, the most prominent alteration seen in the retina was abnormal cone synapses, whereas such comparable synapse changes have not been reported in either rods or cones in other retinal dystrophies, including retinitis pigmentosa.49 This suggests that synaptic abnormalities may be a more common disease mechanism in CORD than other retinal disorders.
Acknowledgments
The work was supported by a grant from the Guide Dogs for the Blind Association. We are also grateful to the patients who kindly agreed to take part in this study.
Abbreviations
AF, autofluorescence
BEM, bull’s eye maculopathy
CORD, cone-rod dystrophies
cSLO, confocal scanning laser ophthalmoscope
EOG, electro-oculogram
ERG, electroretinogram
FFA, fundus fluorescein angiography
PERG, pattern ERG
RPE, retinal pigment epithelium
REFERENCES
- 1.Warburg M, Sjo O, Tranebjaerg L, et al. Deletion mapping of a retinal cone-rod dystrophy: assignment to 18q211. Am J Med Genet 1991;39:288–93. [DOI] [PubMed] [Google Scholar]
- 2.Evans K, Fryer A, Inglehearn C, et al. Genetic linkage of cone-rod retinal dystrophy to chromosome 19q and evidence for segregation distortion. Nat Genet 1994;6:210–13. [DOI] [PubMed] [Google Scholar]
- 3.Freund CL, Gregory-Evans CY, Furukawa T, et al. Cone-rod dystrophy due to mutations in a novel photoreceptor-specific homeobox gene (CRX) essential for maintenance of the photoreceptor. Cell 1997;91:543–53. [DOI] [PubMed] [Google Scholar]
- 4.Cremers FP, van de Pol DJ, van Driel M, et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum Mol Genet 1998;7:355–62. [DOI] [PubMed] [Google Scholar]
- 5.Kelsell RE, Gregory-Evans K, Payne AM, et al. Mutations in the retinal guanylate cyclase (RETGC-1) gene in dominant cone-rod dystrophy. Hum Mol Genet 1998;7:1179–84. [DOI] [PubMed] [Google Scholar]
- 6.Udar N, Yelchits S, Chalukya M, et al. Identification of GUCY2D gene mutations in CORD5 families and evidence of incomplete penetrance. Hum Mutat 2003;21:170–1. [DOI] [PubMed] [Google Scholar]
- 7.Johnson S, Halford S, Morris AG, et al. Genomic organisation and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7). Genomics 2003;81:304–14. [DOI] [PubMed] [Google Scholar]
- 8.Khaliq S, Hameed A, Ismail M, et al. Novel locus for autosomal recessive cone-rod dystrophy CORD8 mapping to chromosome 1q12–q24. Invest Ophthalmol Vis Sci 2000;41:3709–12. [PubMed] [Google Scholar]
- 9.Danciger M, Hendrickson J, Lyon J, et al. CORD9 a new locus for arCRD: mapping to 8p11, estimation of frequency, evaluation of a candidate gene. Invest Ophthalmol Vis Sci 2001;42:2458–65. [PubMed] [Google Scholar]
- 10.Demirci FY, Rigatti BW, Wen G, et al. X-linked cone-rod dystrophy (locus COD1): identification of mutations in RPGR exon ORF15. Am J Hum Genet 2002;70:1049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mears AJ, Hiriyanna S, Vervoort R, et al. Remapping of the RP15 locus for X-linked cone-rod degeneration to Xp11.4–p21.1, and identification of a de novo insertion in the RPGR exon ORF15. Am J Hum Genet 2000;67:1000–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jalkanen R, Demirci FY, Tyynismaa H, et al. A new genetic locus for X linked progressive cone-rod dystrophy. J Med Genet 2003;40:418–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakazawa M, Kikawa E, Chida Y, et al. Autosomal dominant cone-rod dystrophy associated with mutations in codon 244 (Asn244His) and codon 184 (Tyr184Ser) of the peripherin/RDS gene. Arch Ophthalmol 1996;114:72–8. [DOI] [PubMed] [Google Scholar]
- 14.Maugeri A, Klevering BJ, Rohrschneider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone-rod dystrophy. Am J Hum Genet 2000;67:960–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fishman GA, Stone EM, Eliason DA, et al. ABCA4 gene sequence variations in patients with autosomal recessive cone-rod dystrophy. Arch Ophthalmol 2003;121:851–5. [DOI] [PubMed] [Google Scholar]
- 16.Hameed A, Abid A, Aziz A, et al. Evidence of RPGRIP1 gene mutations associated with recessive cone-rod dystrophy. J Med Genet 2003;40:616–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wissinger B, Gamer D, Jagle H, et al. CNGA3 mutations in hereditary cone photoreceptor disorders. Am J Hum Genet 2001;69:722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Downes SM, Holder GE, Fitzke FW, et al. Autosomal dominant cone and cone-rod dystrophy with mutations in the guanylate cyclase activator 1A gene-encoding guanylate cyclase activating protein-1. Arch Ophthalmol 2001;119:96–105. [PubMed] [Google Scholar]
- 19.Kobayashi A, Higashide T, Hamasaki D, et al. HRG4 (UNC119) mutation found in cone-rod dystrophy causes retinal degeneration in a transgenic model. Invest Ophthalmol Vis Sci 2000;41:3268–77. [PubMed] [Google Scholar]
- 20.Sohocki MM, Perrault I, Leroy BP, et al. Prevalence of AIPL1 mutations in inherited retinal degenerative disease. Mol Genet Metab 2000;70:142–150. [DOI] [PubMed] [Google Scholar]
- 21.Krill AE, Deutman AF, Fishman M. The cone degenerations. Doc Ophthalmol 1973;35:1–80. [DOI] [PubMed] [Google Scholar]
- 22.Moore AT. Cone and cone-rod dystrophies. J Med Genet 1992;29:289–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kelsell RE, Gregory-Evans K, Gregory-Evans CY, et al. Localization of a gene (CORD7) for a dominant cone-rod dystrophy to chromosome 6q. Am J Hum Genet 1998;63:274–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y, Okamoto M, Schmitz F, et al. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature 1997;388:593–8. [DOI] [PubMed] [Google Scholar]
- 25.Sun L, Bittner MA, Holz RW. RIM, a component of the presynaptic active zone and modulator of exocytosis, binds 14–3–3 through its N-terminus. J Biol Chem 2003;278:38301–9. [DOI] [PubMed] [Google Scholar]
- 26.Lonart G . RIM1: an edge for presynaptic plasticity. Trends Neurosci 2002;25:329–32. [DOI] [PubMed] [Google Scholar]
- 27.Marmor MF, Zrenner E. Standard for clinical electro-oculography. International Society for Clinical Electrophysiology of Vision. Arch Ophthalmol 1993;111:601–4. [DOI] [PubMed] [Google Scholar]
- 28.Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 update). Doc Ophthalmol 1998;97:143–56. [DOI] [PubMed] [Google Scholar]
- 29.Bach M, Hawlina M, Holder GE, et al. Standard for pattern electroretinography. International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol 2000;101:11–18. [DOI] [PubMed] [Google Scholar]
- 30.Jacobson SG, Voigt WJ, Parel JM, et al. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology 1986;93:1604–11. [DOI] [PubMed] [Google Scholar]
- 31.Steinmetz RL, Haimovici R, Jubb C, et al. Symptomatic abnormalities of dark adaptation in patients with age-related Bruch’s membrane change. Br J Ophthalmol 1993;77:549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alexander KR, Fishman GA. Prolonged rod dark adaptation in retinitis pigmentosa. Br J Ophthalmol 1984;68:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen JC, Fitzke FW, Pauleikhoff D, et al. Functional loss in age-related Bruch’s membrane change with choroidal perfusion defect. Invest Ophthalmol Vis Sci 1992;33:334–40. [PubMed] [Google Scholar]
- 34.Von Rückmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol 1995;79:407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Von Rückmann A, Fitzke FW, Bird AC. Distribution of pigment epithelium autofluorescence in retinal disease state recorded in vivo and its change over time. Graefes Arch Clin Exp Ophthalmol 1999;237:1–9. [DOI] [PubMed] [Google Scholar]
- 36.Von Rückmann A, Fitzke FW, Bird AC. Fundus autofluorescence in age related macular disease imaged with a scanning laser ophthalmoscope. Invest Ophthalmol Vis Sci 1997;38:478–86. [PubMed] [Google Scholar]
- 37.Dorey CK, Wu G, Ebenstein D, et al. Cell loss in the ageing retina: relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci 1989;30:1691–9. [PubMed] [Google Scholar]
- 38.Robson AG, El-Amir A, Bailey C, et al. Pattern ERG correlates of abnormal fundus autofluorescence in patients with retinitis pigmentosa and normal visual acuity. Invest Ophthalmol Vis Sci 2003;44:3544–50. [DOI] [PubMed] [Google Scholar]
- 39.Kurz-Levin M, Halfyard AS, Bunce C, et al. Clinical variations in assessment of bull’s eye maculopathy. Arch Ophthalmol 2002;120:567–75. [DOI] [PubMed] [Google Scholar]
- 40.Holder GE, Robson AG, Hogg CR, et al. Pattern ERG: clinical overview, and some observations on associated fundus autofluorescence imaging in inherited maculopathy. Doc Ophthalmol 2003;106:17–23. [DOI] [PubMed] [Google Scholar]
- 41.Kearns TP, Hollenhorst RW. Chloroquine retinopathy: evaluation by fluorescein angiography. Arch Ophthalmol 1966;76:378–84. [DOI] [PubMed] [Google Scholar]
- 42.Fishman GA, Fishman M, Maggiano J. Macular lesions associated with retinitis pigmentosa. Arch Ophthalmol 1977;95:798–803. [DOI] [PubMed] [Google Scholar]
- 43.Deutman AF. Benign concentric annular macular dystrophy. Am J Ophthalmol 1974;78:384–96. [DOI] [PubMed] [Google Scholar]
- 44.O’Donnell FE, Welch RB. Fenestrated sheen macular dystrophy. A new autosomal dominant maculopathy. Arch Ophthalmol 1979;97:1292–6. [DOI] [PubMed] [Google Scholar]
- 45.Betz A, Thakur P, Junge HJ, et al. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron 2001;30:183–96. [DOI] [PubMed] [Google Scholar]
- 46.Martin TF. Prime movers of synaptic vesicle exocytosis. Neuron 2002;34:9–12. [DOI] [PubMed] [Google Scholar]
- 47.Lloyd TE, Bellen HJ. pRIMing synaptic vesicles for fusion. Nat Neurosci 2001;4:965–6. [DOI] [PubMed] [Google Scholar]
- 48.Koushika SP, Richmond JE, Hadwiger G, et al. A post-docking role for active zone protein Rim. Nat Neurosci 2001;4:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gregory-Evans K, Fariss RN, Possin DE, et al. Abnormal cone synapses in human cone-rod dystrophy. Ophthalmology 1998;105:2306–12. [DOI] [PubMed] [Google Scholar]