

Abstract
Aims: To characterise the detailed phenotype of “cone dystrophy with supernormal rod ERG” in a case series of 10 patients.
Methods: 10 affected patients were examined clinically and underwent colour fundus photography, with nine undergoing detailed electrophysiological testing. Five patients were assessed further with fundus autofluorescence (AF) imaging, automated photopic and dark adapted perimetry, and dark adaptometry. Detailed colour vision assessment was performed in six subjects. Blood samples were taken from four patients for DNA extraction and mutation screening of NR2E3 was undertaken.
Results: The onset of symptoms was in the first and second decades of life. Subjects presented with reduced central vision and marked photophobia. All individuals were myopic and colour vision testing revealed severely reduced colour discrimination predominantly along the red-green axes; tritan colour vision was relatively well preserved. Nyctalopia is a later feature of the disorder. Funduscopy and AF imaging revealed a range of macular appearances. There was electrophysiological evidence of marked macular dysfunction, reduced and delayed cone responses, and supernormal and delayed rod responses. Photopic and dark adapted perimetry revealed central scotomata with widespread peripheral sensitivity loss. No disease causing sequence variants in NR2E3 were identified.
Conclusions: The largest case series to date has been described of the clinical, psychophysical and electrophysiological characteristics of this unusual cone dystrophy with supernormal rod responses. Electrophysiological data were consistent with a post-phototransduction, but pre-inner nuclear layer, site of dysfunction. While the definitive diagnosis can only be made with electrophysiological testing, several characteristics that may increase suspicion of this diagnosis are presented.
Keywords: cone dystrophy, supernormal rod electroretinogram
The cone and cone-rod dystrophies are a clinically and genetically heterogeneous group of retinal disorders.1,2 A range of different phenotypes have been described including an unusual cone disorder associated with supernormal and delayed rod ERG b-waves.3–8 This retinal dystrophy was first described by Gouras et al in two siblings with generalised loss of cone vision, clinical evidence of progression, and nyctalopia.3 Cone ERGs were markedly reduced while the rod b-wave was supernormal in amplitude in response to intense flashes, but smaller than normal and delayed over the lower intensity series. Alexander and Fishman reported two similar cases with supernormal rod ERGs without nyctalopia, suggestive of good rod function despite an abnormal scotopic ERG.4
Supernormal ERGs occur in only one other inherited retinal disease, enhanced S-cone syndrome (ESCS), where responses to short wavelength stimulation are enhanced compared with normal.9,10 In ESCS there is an increase in S-cone numbers related to mutation within NR2E3, a gene believed to play a part in determining cone cell fate.11,12 However, while supernormal rod ERGs can be recorded in the cone dystrophy with supernormal rod responses phenotype (COD/SuperROD), there is no reported enhancement of rod dependent function, and the mechanism of disease is currently unknown. It was initially proposed that defects in phototransduction secondary to dysregulation of intracellular cGMP levels may be involved.3,7,13–15 However, a more recent report has suggested that phototransduction is normal but there may be a delay in the activation of inner nuclear layer activity.5
We have characterised the phenotype of cone dystrophy with supernormal rod responses in detail, to improve our knowledge of the natural history of the disorder, thereby allowing better advice on prognosis and genetic counselling, and with the hope of shedding light upon disease mechanisms. In view of the high amplitude of the electrophysiological responses characteristic of this disorder and the fact that rod photoreceptors share more properties with S-cones than do mid-spectral cones,16,17 mutation screening of NR2E3 was performed in a subset of our affected subjects.
PATIENTS AND METHODS
Patients
Ten subjects (five simplex cases, a sibling pair, and three siblings) of varied ethnic origin, with the phenotype of cone dystrophy with supernormal rod responses (COD/SuperROD) were ascertained electrophysiologically. A detailed medical and ophthalmic history was obtained and a full ophthalmological examination performed. All patients underwent colour fundus photography and five subjects underwent fundus autofluorescence (AF) imaging, automated photopic and dark adapted perimetry, and dark adaptometry. Six individuals had a detailed colour vision assessment. Blood samples were taken from four individuals for DNA extraction and mutation screening of NR2E3.
Methods
Clinical
Fundus autofluorescence imaging was undertaken using the confocal scanning laser ophthalmoscope (cSLO) (Zeiss Prototype; Carl Zeiss Inc, Oberkochen, Germany). Electrophysiological assessment included an electro-oculogram (EOG), full field electroretinogram (ERG) and pattern ERG (PERG), incorporating the protocols recommended by the International Society for Clinical Electrophysiology of Vision.18–20
Colour vision testing included the use of anomaloscopy, the Hardy, Rand, Rittler (HRR) plates (American Optical Company, NY, USA), the enlarged Farnsworth D-15 (PV-16), the standard and enlarged Mollon-Reffin (MR) minimal test,21 and a computerised colour vision test.22,23 For case 4, spectral sensitivity data were obtained. To isolate short wave cones, 1 degree, 500 ms, monochromatic test flashes were presented in Maxwellian view on a 3 degree background. The latter had a wavelength of 520 nm and a troland value of 950: it was designed to elevate the threshold of rods and of any middle or long wave cones. Test wavelengths ranged from 420 to 510 nm, in 15 nm increments. Thresholds were measured by a Yes/No method and stimulus radiance was adjusted by an adaptive threshold procedure under computer control. Wavelengths were tested in random order, and each threshold was measured in two independent runs. To isolate a possible long wave class of cones, 1 degree, 500 ms monochromatic flashes were presented on a 3 degree background of 480 nm and 65 trolands. The test wavelengths were 500, 520, 540, 560, 580, 600, and 650 nm. Procedures were as for the short wave tests.
Five affected subjects underwent detailed perimetry and dark adaptation. Static threshold perimetry in the dark and light adapted states was performed using a Humphrey field analyser (Allergan Humphrey, Hertford, UK).24–26 For dark adapted visual fields, the pupil was dilated with 2.5% phenylephrine hydrochloride and 1% tropicamide, and the patient was dark adapted for 45 minutes. The Humphrey field analyser was modified for use in dark adapted conditions and has been described in detail previously.24–26
For dark adaptometry, two test locations were chosen at 3° and 9°. The Humphrey field analyser used was controlled by a custom computerised program (PS/2 model 50; International Business Machines, Armonk, NY, USA).26,27
NR2E3 mutation screening
Total genomic DNA was extracted from blood samples using a Nucleon Biosciences kit. The coding sequences of NR2E3 were amplified by polymerase chain reaction (PCR) in each individual using primer sequences and conditions as previously published.11 Direct sequencing of PCR products was carried out on an ABI 3100 Genetic Analyser using the original PCR primers in the sequencing reactions.
RESULTS
Clinical
The ethnicity of our 10 subjects was diverse, with geographical origin from Britain, Somalia, Pakistan, Iran, and the United Arab Emirates. The onset of symptoms was in the first and second decades of life. Subjects presented with reduced central vision and marked photophobia that was a very significant source of discomfort and difficulty. Three individuals had mild nystagmus. All subjects were moderately to highly myopic, with variable degrees of astigmatism. Four individuals were aware of a progressive deterioration in visual acuity (VA) and colour vision, and there was progressive reduction in recorded VA in two further patients.
The three oldest patients in the case series complained of nyctalopia, whereas the younger subjects denied difficulties with night vision. This suggests that poor night vision may be a later feature of the disorder. Detailed colour vision testing revealed that all 10 individuals had severely reduced colour discrimination predominantly along the red-green axes with relative sparing of the tritan axis. Spectral sensitivity data obtained in one individual using Maxwellian view are shown in figure 1. Under conditions that isolate short wave cones there is a peak sensitivity in the region of 440 nm. It is clear that the patient retains short wave cones with a normal relative spectral sensitivity; evidence consistent with the preserved tritan discrimination seen in all the patients. These data are consistent with the sibling pair originally described by Gouras et al, in whom relative preservation of S-cone ERGs compared to mid-spectral cone ERG responses was described.3
Figure 1.

Case 4. Spectral sensitivity measurements. The mean thresholds obtained under conditions that isolate short wave cones are shown as solid circles: there is a peak sensitivity in the region of 440 nm. The solid line represents the short wave cone fundamental of Smith and Pokorny.32 It is clear that the patient retains short wave cones with a normal relative spectral sensitivity. The mean thresholds obtained under conditions that isolate a possible long wave class of cones are shown as open circles: there is a peak sensitivity in the region of 560 nm. The solid line represents the long wave cone fundamental of Smith and Pokorny. It is clear that the patient retains at least one class of cone in the long wave region.
Clinical findings are summarised in tables 1 and 2.
Table 1.
Summary of clinical findings
| Case | Sex | Age | Visual acuity RE–LE | Fundus | AF imaging | ERG | OFF responses | PERG | Colour vision |
| 1 | F | 21 | 6/60–6/60 | Bilateral mild macular atrophy with temporal optic nerve pallor (fig 2D) | Bilateral perifoveal rings of relative increased AF (fig 2E) | Reduced and delayed cone responses Supernormal and delayed rod responses | Reduced with delay | ND | Bilateral markedly reduced red/green discrimination with reasonable tritan discrimination |
| 2 | M | 16 | 6/12–6/18 | Bilateral mild macular atrophy with temporal optic nerve pallor | Early bilateral perifoveal rings of relative increased AF | Reduced and delayed cone responses Supernormal and delayed rod responses | Delayed | ND | Bilateral markedly reduced red/green discrimination with reasonable tritan discrimination |
| 3 | F | 37 | 6/24–6/24 | Bilateral macular RPE changes with areas of atrophy and pigmentation | Bilateral central macular area of increased AF with a subtle surrounding ring of increased AF | Reduced and delayed cone responses Supernormal and delayed rod responses | Reduced with delay | Absent | Bilateral reduced red/green discrimination with good tritan discrimination |
| 4 | M | 27 | 6/36–6/60 | Bilateral mild macular RPE disturbance with temporal optic nerve pallor | Bilateral decreased AF centrally with a surrounding ring of relative increased AF | Reduced and delayed cone responses Supernormal and delayed rod responses | Reduced with delay | Absent | Bilateral markedly reduced red/green discrimination with good tritan function |
| 5 | M | 36 | 6/36–6/60 | Bilateral “bull’s eye maculopathy (BEM)-like” RPE changes (fig 2B) | Bilateral central macular area of markedly increased AF surrounded by a ring of relative decreased AF (fig 2C) | Reduced and delayed cone responses Supernormal-like and delayed rod responses | Reduced with delay | Absent | Bilateral markedly reduced red/green discrimination with reasonable tritan discrimination |
| 6 | F | 26 | 6/24–6/24 | Bilateral mild macular atrophy with temporal optic nerve pallor | ND | Reduced and delayed cone responses Supernormal and delayed rod responses | ND | Absent | Bilateral markedly reduced red/green discrimination with reasonable tritan function |
| 7 | F | 25 | 6/36–6/60 | Bilateral macular atrophy with mild temporal optic nerve pallor (fig 2A) | ND | Reduced and delayed cone responses Supernormal-like and delayed rod responses | Delayed | Absent | Bilateral markedly reduced red/green discrimination with reasonable tritan function |
| 8 | M | 26 | 2/36–6/36 | Right old subtotal retinal detachment; Left sided macular atrophy (fig 3) | ND | ND | ND | ND | Bilateral reduced red/green discrimination with reasonable tritan function |
| 9 | F | 18 | 6/60–6/60 | Normal appearance | ND | Reduced and delayed cone responses Supernormal and delayed rod responses | ND | Absent | Bilateral markedly reduced red/green discrimination with good tritan discrimination |
| 10 | M | 13 | 6/18–6/18 | Normal appearance | ND | Reduced and delayed cone responses Supernormal and delayed rod responses | Delayed | Absent | Bilateral markedly reduced red/green discrimination with reasonable tritan function |
Cases 1 and 2 are brother and sister. Cases 6, 7, and 8 are siblings. ND, not done.
Table 2.
Refraction, VA progression, nyctalopia, and perimetry
| Case | Age | Refraction | Presenting VA | Current VA | Night blindness | Photopic and dark adapted perimetry |
| RE | RE | RE | ||||
| LE | LE | LE | ||||
| 1 | 21 | −5.0/−0.50×90 | 6/24 | 6/60 | No | Central and sup>inf widespread peripheral |
| −5.50 DS | 6/24 | 6/60 | VF loss | |||
| 2 | 16 | −3.5/−0.75×50 | 6/12 | 6/12 | No | Central and sup>inf widespread peripheral |
| −4.5/−0.50×130 | 6/12 | 6/18 | VF loss | |||
| 3 | 37 | −4.0/−0.50×90 | 6/24 | 6/24 | Yes | Central and sup>inf widespread peripheral |
| −4.50 DS | 6/24 | 6/24 | VF loss | |||
| 4 | 27 | −4.0/−1.5×180 | 6/36 | 6/36 | Yes | Central and sup>inf widespread peripheral |
| −5.0/−2.0×180 | 6/36 | 6/60 | VF loss | |||
| 5 | 36 | −1.0/−1.5×65 | 6/12 | 6/36 | Yes | Central and sup>inf widespread peripheral |
| −1.0/−2.25×120 | 6/18 | 6/60 | VF loss | |||
| 6 | 26 | −2.5/−1.0×160 | – | 6/24 | No | ND |
| −3.75/−0.5×45 | 6/24 | |||||
| 7 | 25 | −2.0/−1.5×90 | – | 6/36 | No | ND |
| −2.50/−0.5×45 | 6/60 | |||||
| 8 | 26 | −2.0/−0.5×180 | – | 2/36 | No | ND |
| −3.50/−0.5×85 | 6/36 | |||||
| 9 | 18 | −6.25/−3.25×20 | 6/36 | 6/60 | No | ND |
| −7.0/−2.0×165 | 6/36 | 6/60 | ||||
| 10 | 13 | −6.50/−0.25×90 | 6/12 | 6/18 | No | ND |
| −6.50/−0.50×10 | 6/12 | 6/18 |
Sup, superior; Inf, inferior; ND, not done; VF, visual field.
There was a range of macular appearances including normal fundi (two subjects), mild disturbance of the retinal pigment epithelium (RPE), and “bull’s eye” maculopathy (BEM) (table 1, fig 2). Mild pallor of the temporal optic nerve was present in five subjects. The peripheral retina was normal. In one individual (case 8) there was evidence of a unilateral subtotal retinal detachment that had flattened spontaneously (fig 3). In three individuals AF imaging revealed a perifoveal ring of increased AF (table 1, fig 2). In two older subjects an area of increased AF was seen at the central macula; in one of these a perifoveal ring of increased AF was evident, while in the other the central highly fluorescent zone was encircled by an area of relative decreased AF (table 1, fig 2).
Figure 2.
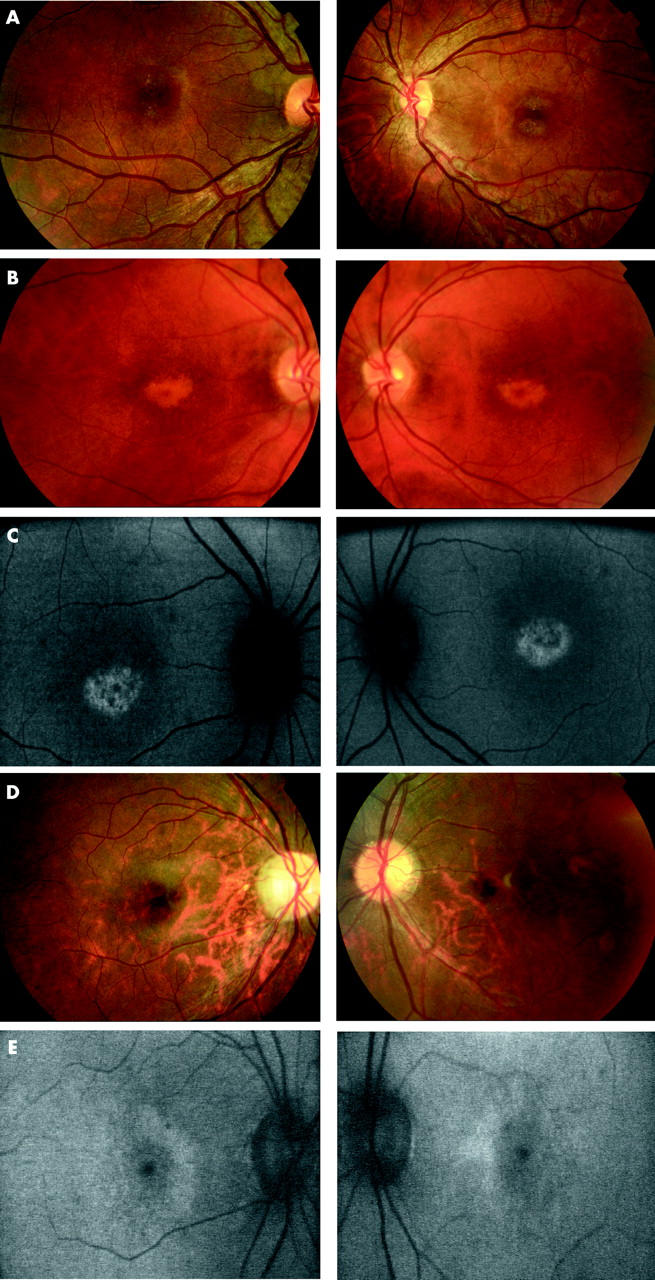
(A) Case 7. Fundus photographs showing bilateral macular atrophy, left worse than right. (B) Case 5. Fundus photographs showing bilateral “bull’s eye maculopathy (BEM)-like” RPE changes. (C) Case 5. Fundus autofluorescence imaging showing bilateral central macular areas of markedly increased AF surrounded by a ring of relative decreased AF. (D) Case 1. Fundus photographs showing bilateral mild macular atrophy with mild temporal optic nerve pallor. (E) Case 1. Fundus autofluorescence imaging showing bilateral perifoveal rings of relative increased AF.
Figure 3.

Case 8. Right sided old subtotal retinal detachment that has flattened spontaneously and left sided macular atrophy.
The PERG was absent in the seven subjects in whom testing was possible. There were reduced and delayed cone responses in all subjects. All nine patients had a characteristic appearance of the standard mixed rod-cone ERG, not seen in other retinal disorders (fig 4), where the a-wave commences normally, but then develops a broadened trough with delay before a high amplitude sharply rising b-wave. In all subjects, rod ERG amplitudes were found to be subnormal and undetectable at the lowest flash energies, but as stimulus intensity increased, an abrupt increase in amplitude was seen, which at the higher flash energies may approach or exceed the upper limit of normal (supernormal) (fig 4). Characteristic of the disorder, was a profound and rapid increase in the amplitude of a delayed b-wave despite only a relatively small increase in stimulus intensity. The marked delay of the rod ERG b-wave is most clearly seen at lower intensities. These patterns of ERG abnormality are consistent with previous reports.3–6 The slope of the ERG a-wave in both cone and rod systems was within normal limits in all patients. Delayed recovery from the peak of the photopic b-wave, with no evidence of an i-wave, was seen in all individuals (fig 4). Of the seven subjects tested, all had delayed OFF responses. Representative electrophysiological traces appear in figure 4.
Figure 4.

Representative rod and cone ERG traces. Rows A–C are from three patients (one eye only), with row D a set of normal traces. Case 1 (row A) aged 17 years; case 2 (row B) aged 12 years; and case 10 (row C) aged 12 years. All three patients had a characteristic appearance of the standard mixed rod-cone ERG, where the a-wave commences normally, but then develops a broadened trough with delay before a high amplitude sharply rising b-wave. In all subjects, rod ERG amplitudes were found to be subnormal and undetectable at the lowest flash energies, but as stimulus intensity increased, an abrupt increase in amplitude was seen, which at the higher flash energies exceeded the upper limit of normal (supernormal). Characteristic of the disorder was a profound and rapid increase in the amplitude of a delayed b-wave despite only a relatively low increase in stimulus intensity. The marked delay of the rod ERG b-wave is most clearly seen at lower intensities. Cone responses were reduced and delayed.
We have obtained electrophysiological data establishing progressive deterioration of retinal function in the one subject in whom repeat testing has been performed (case 4; fig 5). In case 8, electrophysiological testing was not possible, but he had a clinically similar phenotype to his two sisters (cases 6 and 7) who had classic ERG findings and were all part of a highly consanguineous pedigree.
Figure 5.

Representative rod and cone ERG traces of case 4 showing progression over time. Row A shows recordings from the right eye when first seen aged 23; row B when next seen aged 25. The lower traces (C) are a representative normal. The rod ERGs show no definite evidence of deterioration, but there is evidence of progression in the cone ERGs. The flicker ERG adopts an abnormal waveform with further implicit time increase, and the photopic single flash ERG shows clear deterioration with a-wave implicit time increase and both a- and b-wave amplitude deterioration.
There was a history of consanguinity in four families (three simplex cases and the three siblings). In the remaining cases there was no family history of retinal disorders, with cases either being isolated or a sibling pair, thereby making the most likely mode of inheritance autosomal recessive.
Perimetry and kinetics
Five affected individuals underwent more detailed psychophysical investigation. Photopic testing demonstrated decreased central visual field sensitivity in the order of 10–20 dB, with additional peripheral sensitivity loss, most marked in the superior visual field. Dark adapted perimetry revealed central and peripheral sensitivity loss in the order of 15–30 dB, with similar rod and cone threshold elevations. The superior visual field was more affected than the inferior (fig 6). Dark adaptometry showed no clear delay in adaptation; however, there were several patients in whom the rod-cone break was poorly defined.
Figure 6.
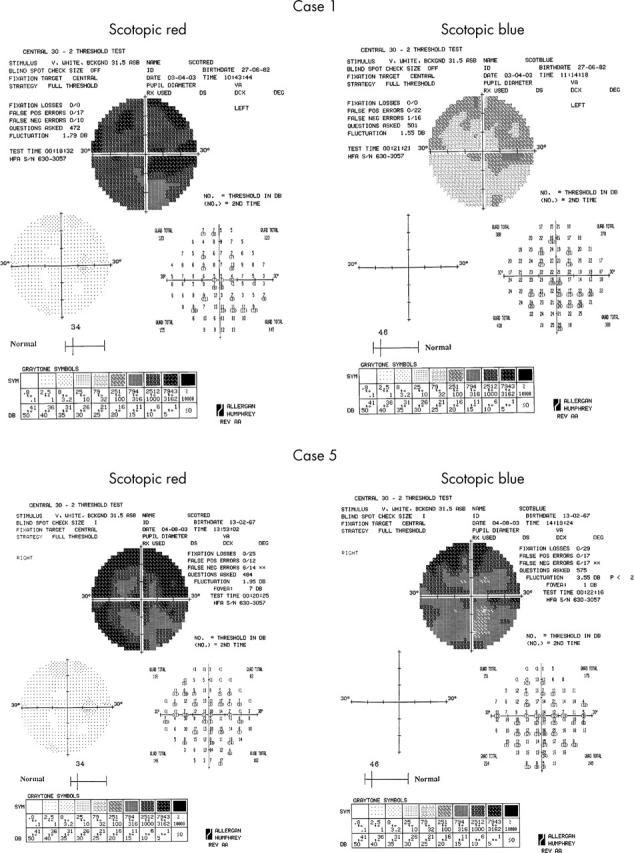
Scotopic red and blue 30° static threshold perimetry of cases 1 and 5, showing central reduction of both cone and rod sensitivities, with peripheral loss being more marked in the inferior retina.
NR2E3 mutation screening
Mutation screening of the nine exons and their flanking splice site regions of NR2E3 failed to identify any disease causing sequence variants in four affected individuals (cases 1–4).
DISCUSSION
We describe the detailed phenotypes of 10 patients with “cone dystrophy with supernormal rod responses” (COD/SuperROD). While ERG recordings are required to make a definitive diagnosis, there are several consistent clinical features in our case series. The retinal dystrophy is characterised by onset in the first or second decades of life, with very marked photophobia, myopia, reduced colour vision along the red-green axis with relatively preserved tritan discrimination, and central scotomata with peripheral widespread sensitivity loss predominating in the superior visual field. Nyctalopia is a later feature of the disorder. There was often RPE disturbance at the macula with a normal retinal periphery. AF imaging demonstrated either a perifoveal ring or a central macular area of relative increased AF. These clinical characteristics may be helpful in suggesting the presence of a COD/SuperROD phenotype.
AF imaging allows the visualisation of the RPE by taking advantage of its intrinsic fluorescence derived from its lipofuscin content.28,29 In three individuals AF imaging revealed a perifoveal ring of increased AF. In the two oldest subjects an area of increased AF was seen at the central macula. The presence of these two AF phenotypes suggests that over time there is a cumulative increase of autofluorescent material at the central macula. This increased lipofuscin is most likely to reflect the inability of the RPE to process outer segment debris, or alternatively increased outer segment turnover. It has been demonstrated histologically that the number of photoreceptor cells is reduced in the presence of increased quantities of lipofuscin in the RPE, leading to the proposal that autofluorescent material may accumulate before cell death.30,31
The PERG was absent in all subjects in whom testing was performed, indicative of marked macular dysfunction. ERG revealed generalised dysfunction of rod and cone systems. The initial phase of both rod and cone a-waves was well formed, suggesting that the kinetics of phototransduction are within normal limits and implying that this disorder is not a primary phototransduction abnormality. The ultimately high amplitude dark adapted ERG b-waves suggest relatively normal inner nuclear layer function. Delayed recovery from the peak of the photopic b-wave, with no evidence of an i-wave, was seen in all individuals and is an unusual finding, the implication of which is unknown. A delayed OFF response recorded is a relatively uncommon finding in retinal dystrophies (GE Holder, unpublished data), and may relate to the undetectable i-wave. The electrophysiological data are consistent with a site of dysfunction that is likely to be post-phototransduction but pre-inner nuclear layer, most probably at the first synapse or alternatively at the horizontal cells. These data are in agreement with those obtained by Hood et al.5 The putative synaptic abnormality would be in both rod and cone systems and may represent a “gated mechanism,” whereby a threshold needs to be exceeded, following which transmission is restored. The period required to reach this threshold may correspond to the broadened and flattened a-wave trough, followed by a rapidly rising, high amplitude b-wave, as previously suggested.5
Evidence of disease progression has been suggested previously on clinical grounds alone.3 In addition to clinical findings of progression, one patient in the current series had progressive deterioration of retinal function evidenced by serial ERG recording.
Colour vision data reported previously are variable and sparse, but in this series, detailed colour vision testing revealed that all affected individuals had severely reduced colour discrimination predominantly along the red-green axes, with far better residual tritan colour vision. Such preservation may increase suspicion of the presence of COD/SuperROD. The explanation for this is unclear but may relate either to a lower susceptibility of S-cones to the disease process than L-/M-cones or, if the site of disease is post-receptoral, to an alternative mode of post-receptoral processing for S-cones not available to L-/M-cones. This latter process may be similar to that proposed for blue cone monochromatism, whereby colour discrimination is derived via a comparison of quantum catches in the S-cones and rods.17
In the absence of any clear link between this unusual retinal disorder and a known retinal process, the identification of the gene mutated in this condition would appear currently to be the only way to elucidate the disease mechanisms. The high prevalence of consanguinity identified in this case series suggests that there may be suitable families for homozygosity mapping to assist in isolating the molecular genetic basis of this condition.
Acknowledgments
The work was supported by a grant from the Guide Dogs for the Blind Association and the Foundation Fighting Blindness. We are also grateful to the patients who kindly agreed to take part in this study.
Abbreviations
AF, autofluorescence
BEM, bull’s eye maculopathy
cSLO, confocal scanning laser ophthalmoscope
EOG, electro-oculogram
ERG, electroretinogram
ESCS, enhanced S-cone syndrome
PERG, pattern ERG
RPE, retinal pigment epithelium
VA, visual acuity
REFERENCES
- 1.Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol 2004;88:291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simunovic MP, Moore AT. The cone dystrophies. Eye 1998;12:553–65. [DOI] [PubMed] [Google Scholar]
- 3.Gouras P, Eggers HM, MacKay CJ. Cone dystrophy, nyctalopia, and supernormal rod responses. A new retinal degeneration. Arch Ophthalmol 1983;101:718–724. [DOI] [PubMed] [Google Scholar]
- 4.Alexander KR, Fishman GA. Supernormal scotopic ERG in cone dystrophy. Br J Ophthalmol 1984;68:69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hood DC, Cideciyan AV, Halevy DA, et al. Sites of disease action in a retinal dystrophy with supernormal and delayed rod electroretinogram b-waves. Vis Res 1996;36:889–901. [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg T, Simonsen SE. Retinal cone dysfunction of supernormal rod ERG type. Acta Ophthalmol 1993;71:246–55. [DOI] [PubMed] [Google Scholar]
- 7.Sandberg MA, Miller S, Berson EL. Rod electroretinograms in an elevated cyclic guanosine monophosphate-type human retinal degeneration. Comparison with retinitis pigmentosa. Invest Ophthalmol Vis Sci 1990;31:2283–7. [PubMed] [Google Scholar]
- 8.Kato M, Kobayashi R, Watanabe I. Cone dysfunction and supernormal scotopic electroretinogram with a high-intensity stimulus. Doc Ophthalmol 1993;84:71–81. [DOI] [PubMed] [Google Scholar]
- 9.Jacobson SG, Marmor MF, Kemp CM, et al. SWS (blue) cone hypersensitivity in a newly identified retinal degeneration. Invest Ophthalmol Vis Sci 1990;31:827–38. [PubMed] [Google Scholar]
- 10.Marmor MF, Jacobson SG, Foerster MH, et al. Diagnostic clinical findings of a new syndrome with night blindness, maculopathy, and enhanced S cone sensitivity. Am J Ophthalmol 1990;110:124–34. [DOI] [PubMed] [Google Scholar]
- 11.Haider NB, Jacobson SG, Cideciyan AV, et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet 2000;24:127–31. [DOI] [PubMed] [Google Scholar]
- 12.Milam AH, Rose L, Cideciyan AV, et al. The nuclear receptor NR2E3 plays a role in human retinal photoreceptor differentiation and degeneration. Proc Natl Acad Sci USA 2002;99:473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pawlyk BS, Sandberg MA, Berson EL. Effects of IBMX on the rod ERG of the isolated perfused cat eye: antagonism with light, calcium or L-cis-diltiazem. Vis Res 1991;31:1093–7. [DOI] [PubMed] [Google Scholar]
- 14.Lipton SA. cGMP and EGTA increase the light-sensitive current of retinal rods. Brain Res 1983;265:41–8. [DOI] [PubMed] [Google Scholar]
- 15.Nicol GD, Miller WH. Cyclic GMP injected into retinal rod outer segments increases latency and amplitude of response to illumination. Proc Natl Acad Sci USA 1978;75:5217–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zrenner E, Gouras P. Blue-sensitive cones of the cat produce a rod-like electroretinogram. Invest Ophthalmol Vis Sci 1979;18:1076–81. [PubMed] [Google Scholar]
- 17.Reitner A, Sharpe LT, Zrenner E. Is colour vision possible with only rods and blue-sensitive cones? Nature 1991;352:798–800. [DOI] [PubMed] [Google Scholar]
- 18.Marmor MF, Zrenner E. Standard for clinical electro-oculography. International Society for Clinical Electrophysiology of Vision. Arch Ophthalmol 1993;111:601–4. [DOI] [PubMed] [Google Scholar]
- 19.Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 update). Doc Ophthalmol 1998;97:143–56. [DOI] [PubMed] [Google Scholar]
- 20.Bach M, Hawlina M, Holder GE, et al. Standard for pattern electroretinography. International Society for Clinical Electrophysiology of Vision. Doc Ophthalmol 2000;101:11–18. [DOI] [PubMed] [Google Scholar]
- 21.Mollon JD, Astell S, Reffin JP. A Minimalist test of colour vision. In: Drum B, Moreland JD, Serra A, eds. Colour vision deficiencies X. Amsterdam: Kluwer Academic Publishers, 1991:59–67.
- 22.Mollon JD, Reffin JP. A computer-controlled colour vision test that combines the principles of Chibret and of Stilling. J Physiol 1989;414:5P. [Google Scholar]
- 23.Regan BC, Reffin JP, Mollon JD. Luminance noise and the rapid determination of discrimination ellipses in colour deficiency. Vis Res 1994;34:1279–99. [DOI] [PubMed] [Google Scholar]
- 24.Jacobson SG, Voigt WJ, Parel JM, et al. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology 1986;93:1604–11. [DOI] [PubMed] [Google Scholar]
- 25.Steinmetz RL, Haimovici R, Jubb C, et al. Symptomatic abnormalities of dark adaptation in patients with age related Bruch’s membrane change. Br J Ophthalmol 1993;77:549–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexander KR, Fishman GA. Prolonged rod dark adaptation in retinitis pigmentosa. Br J Ophthalmol 1984;68:561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen JC, Fitzke FW, Pauleikhoff D, et al. Functional loss in age-related Bruch’s membrane change with choroidal perfusion defect. Invest Ophthalmol Vis Sci 1992;33:334–40. [PubMed] [Google Scholar]
- 28.Von Rückmann A, Fitzke FW, Bird AC. Distribution of fundus autofluorescence with a scanning laser ophthalmoscope. Br J Ophthalmol 1995;79:407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Von Rückmann A, Fitzke FW, Bird AC. Distribution of pigment epithelium autofluorescence in retinal disease state recorded in vivo and its change over time. Graefes Arch Clin Exp Ophthalmol 1999;237:1–9. [DOI] [PubMed] [Google Scholar]
- 30.Von Rückmann A, Fitzke FW, Bird AC. Fundus autofluorescence in age related macular disease imaged with a scanning laser ophthalmoscope. Invest Ophthalmol Vis Sci 1997;38:478–86. [PubMed] [Google Scholar]
- 31.Dorey CK, Wu G, Ebenstein D, et al. Cell loss in the ageing retina: relationship to lipofuscin accumulation and macular degeneration. Invest Ophthalmol Vis Sci 1989;30:1691–9. [PubMed] [Google Scholar]
- 32.Smith VC, Pokorny J. Spectral sensitivity of the foveal cone photopigments between 400 and 500 nm. Vis Res 1975;15:161–71. [DOI] [PubMed] [Google Scholar]