

Abstract
The oxidation of nitric oxide (NO) to nitrate by oxyhemoglobin is a fundamental reaction that shapes our understanding of NO biology. This reaction is considered to be the major pathway for NO elimination from the body; it is the basis for a prevalent NO assay; it is a critical feature in the modeling of NO diffusion in the circulatory system; and it informs a variety of therapeutic applications, including NO-inhalation therapy and blood substitute design. Here we show that, under physiological conditions, this reaction is of little significance. Instead, NO preferentially binds to the minor population of the hemoglobin’s vacant hemes in a cooperative manner, nitrosylates hemoglobin thiols, or reacts with liberated superoxide in solution. In the red blood cell, superoxide dismutase eliminates superoxide, increasing the yield of S-nitrosohemoglobin and nitrosylated hemes. Hemoglobin thus serves to regulate the chemistry of NO and maintain it in a bioactive state. These results represent a reversal of the conventional view of hemoglobin in NO biology and motivate a reconsideration of fundamental issues in NO biochemistry and therapy.
The chemistry of nitric oxide (NO) interactions with Hb has served as a ubiquitous model within the field of NO biochemistry. For example, the oxidative interaction of NO with oxyhemoglobin (oxyHb) to produce nitrate is considered to be the major route of NO catabolism (1–3) as well as a reliable method for assaying NO (4); likewise the unique ability of NO to induce displacement of a trans-imidazole heme ligand has been proposed as key to its activation of guanylyl cyclase (5). In the specific realm of the cardiovascular system, these reactions: are fundamental elements of models for NO diffusion (6, 7); played a crucial role in the identification of endothelium derived relaxing factor (6–9); and inform a variety of therapeutic applications, including NO-inhalation therapy (10, 11) and blood substitute design (12, 13).
Measurements of the rates of these reactions show that the NO-mediated oxidation of oxyHb to methemoglobin (metHb) is kinetically competitive with the binding of NO to unoccupied hemes in Hb—with specific rate constants of 3.7 × 107 M−1 sec−1 and 2.6 × 107 M−1 sec−1, respectively (14–16). The rates of NO oxidation of oxymyoglobin and NO binding to ferrous myoglobin are also very similar (3.4 × 107 M−1 sec−1 vs. 2.5 × 107 M−1 sec−1) (16). Such a rapid route of NO metabolism is, however, difficult to reconcile with mammalian NO production rates (17), which are orders of magnitude too low to sustain physiological NO levels (10 nM–1 μM) (7, 18–20), were NO to be freely consumed in these reactions.‖
Previous studies of the NO oxyHb reaction, however, had been performed with NO concentrations 10-fold greater than protein (16). Under physiological conditions, the concentration ratio is starkly different, with NO concentrations 1,000-fold lower than Hb (20). Moreover, there is always a population of heme sites that are unoccupied. In highly oxygenated Hb, as found in arterial blood, this population is small (≈1%) but is nevertheless in excess of NO. The influence of these vacant hemes, in the physiological situation, cannot be ignored; they might successfully compete for NO with the much larger fraction (≈99%) of oxygen-ligated hemes, if NO binding to hemes in oxyHb were cooperative, that is, if NO addition rates were to increase with increasing oxygen saturation. This possibility has not been raised in previous discussions of NO and Hb chemistry (1–7, 16, 21–24). On the contrary, the demonstrated lack of cooperativity in the binding of NO to deoxyhemoglobin (deoxyHb) (14)—which indicates that the intrinsic NO addition rate constants do not change with NO saturation—implicitly shapes the current perspective. It is important to recognize, however, that these results do not imply that the NO addition rates to oxygenated Hb are similarly independent of the oxygen saturation, and thus cannot be assumed to apply to the physiological situation. In addition to these oxidation and addition reactions, recent studies (20, 25, 26) make it clear that additional reactions, in particular S-nitrosylation, should be considered in any assessment of the chemical interplay of NO and human Hb. The S-nitrosylation reaction assumes particular importance inasmuch as it conserves, rather than consumes, NO bioactivity.
In this article, we discuss the reactions that occur on exposure of Hb to NO at relative concentrations that reflect the physiological situation. We show that the addition of NO to oxyHb takes advantage of the cooperative effects of oxygen binding and thus effectively competes with the oxidation reaction. We further find that at high oxygen saturations, reactions that S-nitrosylate the protein occur to a significant extent. Taken as a whole, these data indicate that the interaction of NO with oxyHb, rather than destroying NO bioactivity as widely misapprehended, acts to preserve it—that Hb very cleverly introduces new chemistry, when oxygen saturation is high, that limits oxidation and channels the NO groups into products that preserve their bioactivity. This picture represents a substantial reversal of the conventional thinking on the chemistry of Hb as it pertains to NO biology and has fundamental implications for the general chemistry of heme-containing proteins.
METHODS
Reaction Product Analysis.
To investigate the reaction of NO with oxyHb, we begin by adopting the conventional viewpoint that: NO consumption involves a competition between the oxidation reaction (Eq. 1) and the adduct-forming addition reaction (Eq. 2); and that the specific rate constants for these reactions, namely kox and kadd, are independent of the degree of oxygen saturation (Y) of the hemes.
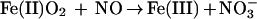 |
1 |
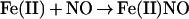 |
2 |
These two assumptions define a perspective of the NO reaction that we refer to as the “simple competition model.” Our analysis of the reaction products, as described in this section, enables us to test the adequacy of this model for describing the chemistry and to recognize and interpret deviations from the behavior implied by it.
In our experiments, NO is introduced as a limiting reagent in an amount substantially smaller than the total amount of oxy- and deoxyhemes. On completion of the reaction, the following relation can be shown to exist among the products:
 |
3 |
in which [Fe(II)]0 and [Fe(II)O2]0 are respectively the initial concentrations of deoxy- and oxyheme. The simple form of Eq. 3 takes advantage of the fact that, independent of Y, at least one of the reactions proceeds under pseudo-first order conditions, and that kox ≈ kadd. The mass balance constraint [Fe(II)NO] + [Fe(III)] = [NO]0 enables us to express the product concentrations relative to the initial concentration of NO, namely [NO]0, as:
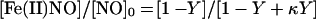 |
4 |
in which κ is kox/kadd, and
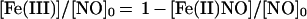 |
5 |
Eqs. 4 and 5 provide the key relationships by which we test the simple competition model—specifically, Eq. 4 indicates that the fractional yield of nitrosylhemoglobin (nitrosylHb) as a function of Y assumes the form of an arc, ranging from 100% yield at Y = 0 to 0% yield at Y = 100%. The degree of curvature of the arc is determined by κ; it is a straight line for κ = 1, but is bowed to one or the other side of this diagonal line as κ is alternatively increased or decreased. For no value of κ does the curve cross the diagonal. It is also worth noting that the derivative of the curve is given by:
 |
6 |
hence as Y → 0, the tangential slope is −kox/kadd, and as Y → 1, it is −kadd/kox. These properties are useful for recognizing possible Y dependences of κ that are inconsistent with the simple model. Similarly, Eq. 5 provides a test for the presence of additional reactions, beyond oxidation (Eq. 1) and addition (Eq. 2): if additional reactions are significant, then [Fe(III)] and [Fe(II)NO] will not account for the total NO ([NO]o) consumed in the reaction, whence [Fe(III)]/[NO]0 + [Fe(II)NO]/[NO]0 < 1.
NO Treatment of Hb.
HbA0 was obtained from Apex Bioscience (Research Triangle Park, NC). Buffer exchange was achieved by dialysis. Deoxygenation was performed by gas exchange with argon in a tonometer. NO was added from a stock solution prepared under ultrapure helium and purified across alkali and cold traps. Stock solutions of NO were prepared in phosphate buffered saline containing 100 μM diethylene triamine pentaacetic acid (DTPA), pH 7.4. NO injections were made via a gas-tight Hamilton syringe with Teflon seal. The concentration of NO in stock solutions was assayed by electrode and by a Sievers 280 NO analyzer (Boulder, CO).
Titration of Normoxic Hb with NO.
Air-oxygenated Hb was titrated with 0.22 μM NO. Samples were analyzed immediately after NO addition by UV-visible spectrophotometry; time between additions varied from 3 to 5 min.
Measurements of S-Nitroso-, Iron Nitrosyl-, and Met-Hb.
Nitroso/nitrosyl derivatives of Hb were measured by using a photolysis-chemiluminescence technique [6-fold excess HgCl2 over protein was added to displace S-nitrosothiol (SNO) (26)]. Samples were kept on ice for a period of 5 min to 2 hr before analyses. MetHb was monitored by UV-visible spectroscopy as the difference absorption above the linear baseline (600–700 nm), and by EPR (below).
EPR Analysis.
EPR spectroscopy was carried out with samples in 4-mm i.d. fused silica tubes, at 76° K, on a Varian E-9 spectrometer. UV-visible spectra were taken after NO addition. The sample was then placed in a deoxygenated EPR tube and plunged into liquid N2. EPR spectra of nitrosylHb or dinitrosyl iron complexes (DNICs) were recorded in a single 4-min scan over 400 G on a Varian E-9 spectrometer operating at 9.274 GHz, with 10-mW microwave power, 10–20 G amplitude of field modulation at 100 kHz, and time constant of 0.250 sec. Spectra of high-spin metHb were recorded with a scan of 1,000 G, 20 G modulation amplitude, time constant of 0.128 sec, under otherwise identical conditions. NitrosylHb was measured by double integration of EPR spectra and by comparison to EPR spectra of Hb(NO)4 standardized with UV-visible spectroscopy. The reproducibility of nitrosylHb measurements was estimated to be ±6% by repeated trials.
Measurement of Oxygen Saturation.
Oxygen saturation of Hb was verified by UV-visible spectroscopy by using a 1-mm anaerobic cuvette.
Extinction Coefficients.
The extinction coefficient spectra of metHb, deoxyHb, iron nitrosylHb, and oxyHb were generated from pure solutions of each species. HbA was diluted into PBS (pH 7.4) to a known final heme concentration [as calculated by the pyridine-hemochromagen method (23)]. MetHb was synthesized by adding excess K3FeCN6. DeoxyHb was measured after the addition of dithionite, and nitrosyl- and oxyHb were measured after saturation with each ligand.
Modeling of UV-Visible Difference Spectra.
Difference spectra were obtained by subtracting the UV-visible spectrum of a given sample before the addition of NO from those after. The simple competition model discussed above predicts that such difference spectra could be approximated by a linear combination of two standard difference spectra: an oxyHb minus metHb spectrum, which gauges the progress of the NO oxidation reaction, and a deoxyHb minus iron nitrosylHb, which gauges the NO addition reaction; the sum of the combining coefficients is fixed by the mass balance ([NO]0). Standard difference spectra were obtained from UV-visible spectra of authentic samples of metHb, oxyHb, nitrosylHb, and deoxyHb. We determined combining coefficients by a least-squares fitting procedure. Inasmuch as the deoxy- and/or oxyheme concentrations can decline during the competition, an additional component (deoxyHb minus oxyHb) could be expected.
RESULTS AND DISCUSSION
EPR spectroscopy was used to assess the formation of nitrosyl heme on addition of NO to Hb preparations with oxygen saturations (Y) in the range 0–80% (typical EPR spectra are shown in Fig. 1A and B). EPR signal intensities were used to quantify the proportion of nitrosylated hemes relative to the NO initially added; the results of this quantification are plotted vs. Y in Fig. 1C (10 mM phosphate) and D (100 mM phosphate). The data obtained at high phosphate levels follow the behavior described by Eq. 4, the solid curves through the data points are graphs of Eq. 4 with κ values for the two depicted curves averaging 1.40 ± 0.06. The data obtained at the lower phosphate level, however, exhibit a notable deviation from the simple model: they cross the diagonal, thus showing a progressive overproduction of nitrosyl heme. Furthermore, the limiting tangential slopes indicate that κ is decreasing with increasing Y. By empirical curve fitting, we found that the data in Fig. 1C are well described by a function of the form (1 + Y)/(1 + cY) (c, a constant). (The solid lines in Fig. 1C are graphs of this function with least-squares best values of the parameter c). This functional form can be assimilated to that of Eq. 4, provided κ is allowed to vary with Y [specifically, κ = (c − 1)(1 − Y)/(1 + Y)]. This result indicates that over the 0 to 80% range of oxygen saturation, κ decreases 7-fold and suggests, by extrapolation, a 100-fold decrease at 90% saturation. We attribute this variation in κ primarily to an increase in kadd,, as kox does not vary by more than a factor of 2, as judged from the limiting slopes and the literature values for kox (Y = 100%) and kadd (Y = 0%).
Figure 1.

Production of iron nitrosylHb by addition of NO to variously oxygenated Hb. (A) EPR spectra of iron-nitrosyl Hb derivatives formed by incubation of 19 μM NO with 393 μM Hb at various degrees of oxygen saturation in 10 mM phosphate buffer, pH 7.4. The oxygen saturations for the largest through smallest EPR signals are 5.5, 32, 50, and 69%, respectively. Spectra show predominantly six coordinate α and β nitrosyl hemes, as typically observed for Hb in R state. (B) EPR spectra of iron-nitrosyl Hb derivatives formed by incubation of 55 μM NO with 380 μM Hb at various degrees of oxygen saturation in 100 mM phosphate, pH 7.4. The oxygen saturations for the largest through smallest EPR signals are 1, 15, 41, 60, and 80%, respectively. Spectra show a significant component of five coordinate α nitrosyl hemes (triplet structure) associated with Hb in T state. (C) Trials conducted with Hb in 10 mM phosphate, pH 7.4. The symbols are experimental results and the solid lines represent a best fit to the functional form for cooperative NO binding. Open diamonds, 393 μM Hb incubated with 19 μM NO; open circles, 350 μM Hb incubated with 15 μM NO plus 0.05% borate (added to bring the buffer concentration to 100 mM as in D); open squares, 365 μM Hb incubated with 15 μM NO and 1,190 units/ml SOD. (D) Trials conducted with Hb in 100 mM phosphate, pH 7.4. The symbols are experimental results, and the lines represent a best fit to the functional form for simple competition between oxidation and NO addition reactions (Eq. 4). Filled circles, 380 μM Hb incubated with 55 μM NO; filled squares, 375 μM Hb incubated with 7 μM NO. Application of the simple competition function to data of C or the cooperativity function to data of D gives an order of magnitude increase in χ2.
We also assessed the formation of oxidized ferric hemes with EPR (data not shown) and UV-visible difference spectroscopy (Fig. 2). The results obtained from samples in 100 mM phosphate conform to the simple competition model: the dashed lines in the figure, which are calculated from the curves depicted in Fig. 1D following Eq. 5, agree extremely well with the experimental measurements. Experiments conducted in 10 mM phosphate, however, show a stark deviation from the simple model behavior. Qualitatively, the results show that heme oxidation never grossly exceeds heme nitrosylation. Moreover, there is a progressive shortfall in the Fe(III) and Fe(II)NO products. This shortfall is indicated by the departure of the experimental points (10 mM phosphate) in Fig. 2, from the curves calculated from the curves depicted in Fig. 1D following Eq. 5, and amounts to as much as ≈20% of added [NO]0. This behavior strongly suggests the presence of additional NO reaction pathways.
Figure 2.

Production of metHb by reaction of NO is disfavored with increasing oxygen saturation. The samples used in Fig. 1 were assayed for metHb production by UV-visible difference spectroscopy. The data are normalized to added [NO]. As in Fig. 1, open diamonds, 10 mM phosphate; open circles, 10 mM phosphate plus borate; open squares, 10 mM phosphate plus SOD; filled circles and filled squares, 100 mM phosphate. The dotted (10 mM phosphate) and dashed (100 mM phosphate) lines are calculated by using Eq. 5 and Fe(II)NO yields in Fig. 1 C and D, respectively. Data show metHb to be disfavored in low phosphate, particularly at high oxygen saturation. Deviations of the data points below the curves suggest the presence of additional reactions for NO. Systematic deviations are most pronounced in low phosphate at high oxygen saturation—i.e., under physiological conditions.
In summary, we find that NO binding to oxyHb is cooperative; that oxidation to ferric heme (metHb) is limited under physiological conditions; and that additional chemistry is occurring in the more oxygenated Hb species that are prevalent in vivo. Our findings might seem at odds with previous literature suggesting that NO binding to Hb is noncooperative (14). The proper conclusion to draw from these prior studies, however, is that NO (ligand) binding to nitrosylHb shows little cooperativity with varying NO-saturation—a scenario of little physiological relevance, because NO is never the dominant ligand in vivo. Our results reflect the physiological situation in which the ligand, NO, binds to Hb with some degree of oxygen saturation. The functional behavior in this situation is, not surprisingly, cooperative. In this regard, experiments of particular interest are those conducted in the presence of high phosphate concentrations (100 mM), which perturb the allosteric modulation of ligand affinity by disfavoring the relaxed [R (oxy)] structure among the partially ligated hemoglobins, as evidenced by the hyperfine structure in the EPR (Fig. 1B) (27). Thus, normal T (deoxy)/R (oxy) interconversion in Hb appears to be essential for “normal” NO function (Fig. 1 A and C). Taken together, the EPR results demonstrate that when the oxygen-induced allosteric transition is unhindered, NO binding to oxygenated Hb is cooperative—a situation that leads to enhanced iron nitrosyl- and limited metHb formation.
To extend these results to arterial oxygen saturation (of ≈99%) and physiological NO concentrations (≈1 μM), we used photolysis–chemiluminescence to measure nitrosyl derivatives of Hb (Fig. 3A). In these experiments, normoxic Hb is in excess of NO, but NO is in excess of the vacant hemes, a scenario disfavoring NO addition. Our results show that even at high oxygen saturation, a substantial fraction of the NO—rather than forming nitrate by the oxidation reaction—forms chemiluminescence-detectable nitrosyl derivatives. Of further interest, the yield of nitrosyl species increases with increasing [Hb] up to a maximum of approximately 50% of NO added (relative to the [Hb] the nitrosyl yield varies from 3 to 0.6%) (Fig. 3A). In the simple competition model, the fraction of nitrosylation products would be independent of protein concentration. These results thus clearly demonstrate that additional reactions, beyond NO binding to vacant hemes to form the nitrosyl-heme derivative, are occurring under these conditions.
Figure 3.

NO addition under normoxic conditions (≈99% O2 saturation) produces nitrosylated Hb. (A) Nitrosyl content of oxyHb (10 mM phosphate/100 μM DTPA, pH 7.4) after exposure to 1.2 μM NO, as measured by photolysis-chemiluminescence (26). Nitrosyl yield increases as a function of Hb concentration (P < 0.05). Solid symbols, absolute yield of NO bound to Hb (FeNO plus SNO); open symbols, percentage of NO added. Data shown are the average of 7 to 19 experiments ±SE. (B) Standard difference spectra of metHb (solid line), deoxyHb (dotted line), and iron nitrosylHb (dashed line) vs. oxyHb (see Materials and Methods for conditions). (C) Difference spectra generated from the exposure of NO to normoxic (≈99% oxygen sat.) Hb. NO was added (in 10 aliquots totaling 2.2 μM) to 33 μM Hb in 100 mM phosphate (solid line) or 10 mM phosphate (dotted line) or 10 mM phosphate plus 0.05% borate (dashed line). Notably, the spectrum in 100 mM phosphate shows the formation of metHb (e.g., peak at 630 nm; see B for comparison); the spectrum in 10 mM phosphate shows formation of iron nitrosyl Hb and some metHb [e.g., peak at 595 nm (nitrosyl) and small peak at 630 nm (met); see B for comparison]; and the spectrum in 10 mM phosphate plus borate shows predominantly iron nitrosylHb (e.g., peak at 595 nm; see B for comparison). (D) Calculated fits for difference spectra shown in C, demonstrating simple (noncooperative) competition between NO binding and oxidation reactions in high phosphate (solid line, 95% metHb) and cooperative binding in low phosphate (dotted line, 54% iron nitrosylHb; only 50% of the added NO accounted for) and low phosphate plus borate (dashed line; 85% iron nitrosylHb). Specifically, spectra in C were fitted, by a least-squares process, to either the simple competition model or the cooperativity model without a mass balance constraint.
To gain further insight into this chemistry, we used the discriminating power of difference absorption spectroscopy. Difference spectra obtained by titration of submicromolar concentrations of NO against 33 μM Hb in room air (99% O2 saturation) are shown in Fig. 3C; standard difference spectra of authentic met-, deoxy-, and nitrosylHb relative to oxyHb are shown in Fig. 3B. If the chemistry were to proceed according to the simple model, then at Y = 99% the oxidation reaction would predominate and the observed difference spectra would closely resemble the metHb minus oxyHb standard difference spectrum. This behavior was observed only at high phosphate concentrations (Fig. 3 C and D), consistent with the EPR results above. At low phosphate concentrations, we found that the difference spectra point largely toward the formation of nitrosylated heme: much of the difference spectrum can be accounted for by the deoxyHb minus nitrosylHb standard spectrum (Fig. 3C). To produce adequate difference-spectrum simulations, it was necessary to include a deoxyHb minus oxyHb component (presumably reflecting compensation for the nitrosylative loss of vacant hemes), and, most significantly, to relax the mass-balance constraint: a measurable fraction of [NO]o was not accounted in the Hb spectra (Fig. 3 C and D); the spectra account for only 50% in low phosphate and 80% in low phosphate plus borate. Taken as a whole, these data extend to normoxic conditions the conclusions made above, namely: direct oxidation by NO is not the predominant reaction at low NO to heme ratios; addition of NO to vacant hemes remains competitive; and further reaction pathways, beyond oxidation and addition, must be occurring.
One additional species that could compete for NO is superoxide—liberated by the autooxidation of oxygenated Hb (28). To examine this possibility, we repeated the experiments detailed in Fig. 3A in the presence of superoxide dismutase (SOD) (Fig. 4A). At all concentrations of Hb used, the presence of SOD increased the yield of Hb nitrosyl derivatives (i.e., total NO bound) to approximately 100% of [NO]o (Fig. 4A). Similarly, SOD led to increases in the yield of nitrosylated hemes detected in the EPR experiments (Fig. 1C). Evidently, under these conditions, superoxide is a significant competitor for NO, or perhaps SOD alters the reactivity (oxidation and/or ligand binding) of Hb. Interestingly, when these experiments were performed with stroma-free Hb, a RBC preparation that contains normal levels of SOD, similar results were observed (Fig. 4A). It is further notable that analogous effects on the nitrosyl yield—assessed by EPR, chemiluminescence, and difference spectroscopy—were obtained when borate was included in the buffer medium (Figs. 1A, 2, and 3C). Borate most likely exerts this effect by altering the ligand on-rate for NO or the reactivity of the oxygen ligand with NO, or perhaps the intrinsic autooxidation rate of Hb. Phosphate levels may also influence these parameters.
Figure 4.

S-nitrosoHb and iron nitrosylHb formed under various physiological air-oxygenated conditions. (A) SOD increases the yield of NO bound to Hb. The experiments in Fig. 3A were repeated in the absence (solid line; 1.2 μM NO) or presence (dashed line; 1.5 μM NO) of 1,190 units/ml of SOD, which enhances the yield of nitrosyl species to approximately 100% of the NO added. Similar nitrosyl yields were obtained by using stroma-free Hb (25 μM), which is enriched in endogenous SOD (open circle). Data shown are the average of five to nine experiments ±SE. (B) EPR spectrum of a DNIC formed by exposure of oxyHb (≈99% sat; 3.93 mM) to NO (36 μM). (C) S-nitrosoHb and iron nitrosylHb formed by exposure of oxyHb (≈99% sat., 48 μM) to NO (1.2 μM). SNO (hatched bar) and FeNO (solid bar) were measured by photolysis-chemiluminesence (26). Data shown are the average of 12 experiments ±SE. (D) Measurement of intraerythrocytic S-nitrosoHb and iron nitrosylHb formed by exposure of oxygenated RBCs (mean [Hb], 25 μM) to 1 μM NO. Isolation of Hb and measurements were as previously described (26). Data are the mean of 12 experiments ±SE.
An important clue to additional reaction pathways comes from our analyses under normoxic conditions: nitrosyl yields as high as 6% of Hb were observed by photolysis-chemiluminesence, notwithstanding the fact that the proportion of heme vacancies is only ≈1%. These nitrosyl species, moreover, did not affect the UV-visible spectra. EPR of samples under these conditions exhibit spectra similar to the DNICs exhaustively studied by Vanin (29), albeit they account for a small percentage of NO added (Fig. 4B). DNICs are known to form from SNOs with which they exist in equilibrium (29). Indeed, chemiluminesence analysis of the products formed on addition of 1.2 μM NO to 48 μM oxyHb, which produces nitrosyl yields of approximately 500 nM (Figs. 1A and 3A), show that ≈80% of this nitrosyl yield is SNO (Fig. 4C). Moreover, treatment of aerated RBCs with physiological concentrations of NO (1 μM) resulted in relatively high yields of intracellular S-nitroso-oxyHb (Fig. 4D). Specifically, analyses revealed (after inherent time delays of ≈30 min) yields of intracellular S-nitrosoHb, iron-nitrosylHb, and metHb of 103 ± 38 nM, 42 ± 15 nM, and 0 nM (i.e., none detectable), respectively (n = 12), and the further appearance of nitrosyl heme adducts on lowering of the oxygen tension, in general agreement with studies on isolated Hb (Figs. 1, 3A, and 4 A and C).
Although the oxidation reaction (Eq. 1) has been given great significance in NO biology, our data demonstrate that it is likely to be of little significance under normal physiological conditions. Because of the low concentration of NO relative to Hb, vacant hemes are in excess over NO. This excess, together with the cooperativity of ligand binding in oxyHb, enables the addition of NO to heme to compete with the oxidation reaction even at high oxygen saturation. Moreover, in oxygenated Hb, additional reactive pathways that preserve NO bioactivity are available, including the production of SNO and DNIC. These results are in keeping with in vivo observations of Hb nitrosyl derivatives, the levels of which are generally unrelated to metHb concentration (27), directly responsive to NO administration (27), and dynamically controlled by allosteric state of Hb (26, 30) but otherwise unrelated to Hb oxygen saturation (26, 27, 30). These findings also help reconcile NO biochemistry with NO production rates in mammals (17)—which are orders of magnitude too low to sustain physiological NO levels, were the oxyHb reaction dominant. In addition, they rationalize the ability of inhaled NO, which is purportedly inactivated by Hb in the lungs (10, 11, 31), to lower systemic blood pressure (11), increase aortic tissue cGMP levels (32), avert sickling of RBCs (33), improve blood flow to ischemic tissues (34), and increase glomerular filtration rate (35).
These discoveries have a strong bearing both on the way NO–heme interactions are modeled and on our understanding of NO biology. The current view of the NO interaction with Hb in vivo is derived from a model in which the elimination as NO3− is dominant and NO release from Hb is inconsequential (1, 3, 4, 6, 7, 15, 16, 36). In reality, Hb musters additional reaction pathways to keep the balance in favor of maintaining the NO group in a bioactive state. These chemical reactions with thiols, metals, and superoxide are the essential elements of the extended paradigm of NO biochemistry presented some years ago (37).
Our results have important implications for rational design of blood substitutes, NO scavengers, and therapeutic NO donors. Additionally, they predict that measurements of NO with the oxyHb assay will tend to underestimate NO production, unless appropriate precautions are taken, and more generally point to limitations of Hb-based approaches for identification of NO bioactivity. Finally, these findings raise fundamental questions. For example, nitrate remains the major metabolic product of NO in vivo, but the question now arises as to its source. It is tempting to suggest the involvement of a heme protein that can neither enforce the cooperativity of ligand binding, nor recruit the thiol reaction pathway. These properties are exemplified in the bacterial flavohemoglobin whose recently identified enzymatic function involves the oxidation of NO to nitrate (38, 39). Whereas the primordial bacterial Hb is designed to metabolize NO (39), mammalian Hb is designed to secure and deliver it (20, 25, 26). These observations suggest that the molecular evolution of Hb was impacted by its NO-related functions.
Acknowledgments
This work was supported by grants (HL52529 and HL59130) from the National Institutes of Health, the Amyotrophic Lateral Sclerosis Association, and MONTS (National Science Foundation—Experimental Program to Stimulate Competitive Research).
ABBREVIATIONS
- SNO
S-nitrosothiol
- oxyHb
oxyhemoglobin
- metHb
methemoglobin
- deoxyHb
deoxyhemoglobin
- nitrosylHb
nitrosylhemoglobin
- DNIC
dinitrosyl iron complex
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
The measured NO synthesis rate is 1.3 millimol per day for the average person (17). To maintain a basal NO concentration of 10 nm—1 μM in vivo (6, 7, 18–20), 130 to 13,000 mol of NO would be consumed per day in reaction with Hb (assuming kox = 3.7 × 107 M−1 sec−1 (14), 5 L vascular volume). The hypothetical NO consumption rate, therefore, is 105- to 107-fold greater than the actual production rate.
References
- 1.Kelm M, Yoshida K. In: Metabolic Fate of Nitric Oxide and Related N-Oxides. 1st Ed. Feelisch M, Stamler J S, editors. London: Wiley; 1996. pp. 47–58. [Google Scholar]
- 2.Pietraforte D, Mallozzi C, Scorza G, Minetti M. Biochemistry. 1995;34:7177–7185. doi: 10.1021/bi00021a032. [DOI] [PubMed] [Google Scholar]
- 3.Wennmalm A, Benthin G, Petersson A S. Br J Pharmacol. 1992;106:507–508. doi: 10.1111/j.1476-5381.1992.tb14365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feelisch M, Kubitzek D, Werringloer J. In: The Oxyhemoglobin Assay. 1st Ed. Feelisch M, Stamler J S, editors. London: Wiley; 1996. pp. 455–478. [Google Scholar]
- 5.Traylor T G, Sharma V S. Biochemistry. 1992;31:2847–2849. doi: 10.1021/bi00126a001. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, Miller M J, Joshi M S, Sadowska-Krowicka H, Clark D A, Lancaster J R., Jr J Biol Chem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 7.Lancaster J R., Jr Proc Natl Acad Sci USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palmer R M, Ferrige A G, Moncada S. Nature (London) 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 9.Ignarro L J, Buga G M, Wood K S, Byrns R E, Chaudhuri G. Proc Natl Acad Sci USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roissant R, Falke F J, Lopez F, Slama K, Pison U, Zapol W M. New Engl J Med. 1993;328:399–405. doi: 10.1056/NEJM199302113280605. [DOI] [PubMed] [Google Scholar]
- 11.Wessel D L, Adatia I, Giglia T M, Thompson J E, Kulik T J. Circulation. 1993;88:2128–2138. doi: 10.1161/01.cir.88.5.2128. [DOI] [PubMed] [Google Scholar]
- 12.Alayash A I, Cashon R E. Mol Med Today. 1995;1:122–127. doi: 10.1016/s1357-4310(95)80089-1. [DOI] [PubMed] [Google Scholar]
- 13.Doherty D H, Doyle M P, Curry S R, Vali R J, Fattor T J, Olson J S, Lemon D D. Nat Biotechnol. 1998;16:672–676. doi: 10.1038/nbt0798-672. [DOI] [PubMed] [Google Scholar]
- 14.Cassoly R, Gibson Q H. J Mol Biol. 1975;91:301–313. doi: 10.1016/0022-2836(75)90382-4. [DOI] [PubMed] [Google Scholar]
- 15.Doyle M P, Hoekstra J W. J Inorg Chem. 1981;14:351–358. doi: 10.1016/s0162-0134(00)80291-3. [DOI] [PubMed] [Google Scholar]
- 16.Eich R F, Li T, Lemon D D, Doherty D H, Curry S R, Aitken J F, Mathews A J X J K, Smith R D, Phillips G N, Jr, Olson J S. Biochemistry. 1996;35:6976–6983. doi: 10.1021/bi960442g. [DOI] [PubMed] [Google Scholar]
- 17.Castillo L, Beaumier L, Ajami A M, Young V R. Proc Natl Acad Sci USA. 1996;93:11460–11465. doi: 10.1073/pnas.93.21.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pinsky D J, Patton S, Mesaros S, Brovkovych V, Kubaszewski E, Grunfeld S, Malinski T. Circ Res. 1997;81:372–379. doi: 10.1161/01.res.81.3.372. [DOI] [PubMed] [Google Scholar]
- 19.Vallance P, Patton S, Bhagat K, MacAllister R, Radomski, Moncada S, Malinski T. Lancet. 1995;346:153–154. doi: 10.1016/s0140-6736(95)91211-8. [DOI] [PubMed] [Google Scholar]
- 20.Jia L, Bonaventura C, Bonaventura J, Stamler J S. Nature (London) 1996;380:221–226. doi: 10.1038/380221a0. [DOI] [PubMed] [Google Scholar]
- 21.Marletta M A, Tayeh M A, Hevel J M. Biofactors. 1990;2:219–225. [PubMed] [Google Scholar]
- 22.Moore E G, Gibson Q H. J Biol Chem. 1976;251:2788–2794. [PubMed] [Google Scholar]
- 23.Antonini E, Brunori M. In: Frontiers in Biology. Neuberger A, Tatum E L, editors. Vol. 21. Amsterdam: North-Holland; 1971. p. 13. [Google Scholar]
- 24.Sharma V S, Traylor T G, Gardiner R, Mizukami H. Biochemistry. 1987;26:3837–3843. doi: 10.1021/bi00387a015. [DOI] [PubMed] [Google Scholar]
- 25.Gow A J, Stamler J S. Nature (London) 1998;391:169–173. doi: 10.1038/34402. [DOI] [PubMed] [Google Scholar]
- 26.Stamler J S, Jia L, Eu J P, McMahon T J, Demchenko I T, Bonaventura J, Gernert K, Piantadosi C A. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi Y, Kobayashi H, Tanaka N, Sato T, Takizawa N, Tomita T. Am J Physiol. 1998;274:H349–H357. doi: 10.1152/ajpheart.1998.274.1.H349. [DOI] [PubMed] [Google Scholar]
- 28.Misra H P, Fridovich I. J Biol Chem. 1972;247:6960–6962. [PubMed] [Google Scholar]
- 29.Vanin A F, Malenkova I V, Serezhenkov V A. Nitric Oxide. 1997;1:191–203. doi: 10.1006/niox.1997.0122. [DOI] [PubMed] [Google Scholar]
- 30.Hall D M, Buettner G R, Mathes R D, Gisolfi C V. J Appl Physiol. 1994;77:548–553. doi: 10.1152/jappl.1994.77.2.548. [DOI] [PubMed] [Google Scholar]
- 31.Westfelt U N, Benthin G, Lundin S, Stenqvist O, Wennmalm A. Br J Pharmacol. 1995;114:1621–1624. doi: 10.1111/j.1476-5381.1995.tb14948.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kermarrec N, Zunic P, Beloucif S, Benessiano J, Drouet L, Payen D. Am J Respir Crit Care Med. 1998;158:833–839. doi: 10.1164/ajrccm.158.3.9709097. [DOI] [PubMed] [Google Scholar]
- 33.Head C A, Brugnara C, Martinez-Ruiz R, Kacmarek R M, Bridges K R, Kuter D, Bloch K D, Zapol W M. J Clin Invest. 1997;100:1193–1198. doi: 10.1172/JCI119631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox-Robichaud A, Payne D, Hasan S U, Ostrovsky L, Fairhead T, Reinhrdt P, Kubes P. J Clin Invest. 1998;101:2497–2505. doi: 10.1172/JCI2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Troncy E, Francoeur M, Salazkin I, Yang F, Charbonneau, Leclerc G, Vinay P, Blaise G. Br J Anaesth. 1997;79:631–640. doi: 10.1093/bja/79.5.631. [DOI] [PubMed] [Google Scholar]
- 36.Sharma V S, Ranney H M. J Biol Chem. 1978;253:6467–6472. [PubMed] [Google Scholar]
- 37.Stamler J S, Singel D J, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 38.Gardner P R, Gardner A M, Martin L A, Salzman A L. Proc Natl Acad Sci USA. 1998;95:10378–10383. doi: 10.1073/pnas.95.18.10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hausladen A, Gow A J, Stamler J S. Proc Natl Acad Sci USA. 1998;95:14100–14105. doi: 10.1073/pnas.95.24.14100. [DOI] [PMC free article] [PubMed] [Google Scholar]