

Abstract
Background: Involvement of the eye has been reported in patients with variant Creutzfeldt-Jakob disease (vCJD), but there is disagreement on whether retinal involvement occurs in sporadic Creutzfeldt-Jakob disease (sCJD).
Methods: Western blotting, paraffin embedded tissue blotting, and immunohistochemistry were used to test whether the abnormal form of the prion protein (PrPSc) accumulates to detectable levels in the eye in a case of the most common subtype of sCJD (MM1).
Results: Low levels of PrPSc were detectable in the retina, localised to the plexiform layers of the central retina. PrPSc was not detectable in other ocular tissues.
Conclusions: The abnormal form of the prion protein is present in the retina in the most common sCJD subtype (MM1), albeit at levels lower than those found previously in vCJD and in sCJD of the VV2 subtype.
Keywords: Creutzfeldt-Jakob disease, prion protein, PRNP codon 129 genotype, retina
Creutzfeldt-Jakob disease (CJD) is a human prion disease that occurs in sporadic, acquired, and familial forms. The cause of sporadic CJD (sCJD) is not known with certainty, but the familial forms of CJD are all associated with mutations in the prion protein gene (PRNP). Acquired forms of the disease include variant CJD (vCJD), resulting from human infection with the bovine spongiform encephalopathy agent, and iatrogenic CJD. Iatrogenic CJD has been associated most recently with blood transfusion but also historically with human cadaveric growth hormone treatment, dura mater transplantation, contaminated neurosurgical instruments, and corneal transplantation.1–3 Although there is no epidemiological evidence for other forms of ophthalmic surgery having resulted in CJD transmission between patients, it cannot be ruled out.4
Determining the extent of tissue involvement in the different forms of CJD is an important component of a rational assessment of the risk of iatrogenic CJD transmission associated with a given medical procedure. Infectivity can be assessed directly by animal transmission studies and the eye has previously been shown to harbour infectivity, although the responsible ocular component was not identified.5 An alternative approach is to use the disease associated, abnormal form of the prion protein, PrPSc, as a marker of infectivity. In general, the tissue distribution of PrPSc in vCJD is very much wider than in sCJD, including the lymphoreticular system in addition to the central nervous system.6,7 The two papers that have reported on ocular tissues in vCJD were in agreement that PrPSc is present at readily detectable levels in neurosensory retina and in the optic nerve, but is undetectable in the cornea, lens, vitreous body, and sclera.6,8 However, these same studies did not concur over sCJD. Even with a highly sensitive Western blotting technique, Wadsworth and colleagues6 were unable to detect PrPSc in any ocular component of the eye from the case of sCJD available to them. We found, however, that PrPSc was detectable at levels similar to those found in the brain and distributed uniformly throughout the plexiform layers in the retina from the eye of a patient with sCJD.8 One possible explanation for this discrepancy is the known diversity of clinical and pathological phenotype in sCJD. This depends partly on the PrPSc type present (either type 1 or type 2) and on the methionine/valine polymorphism at codon 129 of the PRNP gene.9 The eye which we previously examined was from a patient who had type 2 PrPSc in the brain and was homozygous for valine at codon 129 of PRNP (that is, VV2). We have recently had the opportunity to readdress this issue by examining both fixed and frozen eye tissues from a 67 year old man referred to the National CJD Surveillance Unit with suspected CJD. Autopsy examination confirmed the diagnosis of sCJD, and the biochemical and genetic tests identified this to be of the most commonly occurring MM1 subtype. Consent was given for tissue retention and research use.
MATERIALS AND METHODS
Western blot analysis for PrPSc was carried out using the method described previously.8 Briefly, frozen tissues were homogenised in a non-denaturing detergent buffer at 10% tissue weight/volume of buffer. The cleared homogenate was digested with 50 μg/ml proteinase K (Merck Biosciences, Beeston, UK) at 37°C for 1 hour. Protease resistant PrPSc was analysed by Western blotting using 10% NuPage bis-tris gels, buffers, and the manufacturer’s instructions (Invitrogen Life Sciences, Paisley, UK). Western blots were probed with the monoclonal antibody 3F4 (Dako, High Wycombe, UK), horseradish peroxidase conjugated anti-mouse secondary antibody, and the signal captured by enhanced chemiluminescence (ECL+) using x ray film (all from Amersham Biosciences, Bucks, UK). In some experiments a modified tissue dispersion step employing a FastPrep tissue homogeniser (Qbiogene, Cambridge, UK) was used in combination with a preparatory step that employs sodium phosphotungstic acid precipitation of PrPSc from a larger volume of tissue homogenate.3,6,10 Paraffin embedded tissue blotting using the monoclonal antibody 3F4 was performed to localise PrPSc in sections of the fixed eye.11 Finer localisation was achieved using immunohistochemistry with the 12F10 monoclonal antibody (IDS Ltd, Tyne and Wear, UK).8
RESULTS
Western blot analysis using the standard method showed that type 1 PrPSc was abundant in the brain (frontal cortex) from this case of sCJD. In contrast, PrPSc was undetectable in a sample of retina (fig 1A). We then re-sampled the ocular tissues and prepared tissue homogenates using a modified tissue dispersion step employing a FastPrep tissue homogeniser. Standard Western blot analysis again failed to detect PrPSc in the retina even on maximal exposure (fig 1B). The remaining FastPrep tissue extracts (500 μl) were then used in a sodium phosphotungstic acid precipitation assay. Using this method to selectively precipitate PrPSc before Western blot analysis, a positive signal was seen in the retina sample (fig 1C). No PrPSc was detectable in the cornea, lens, iris, vitreous body, sclera, or extraocular muscle in the standard assay or in the phosphotungstic acid precipitation experiments. A faint signal was obtained from the sample of optic nerve (data not shown).
Figure 1.
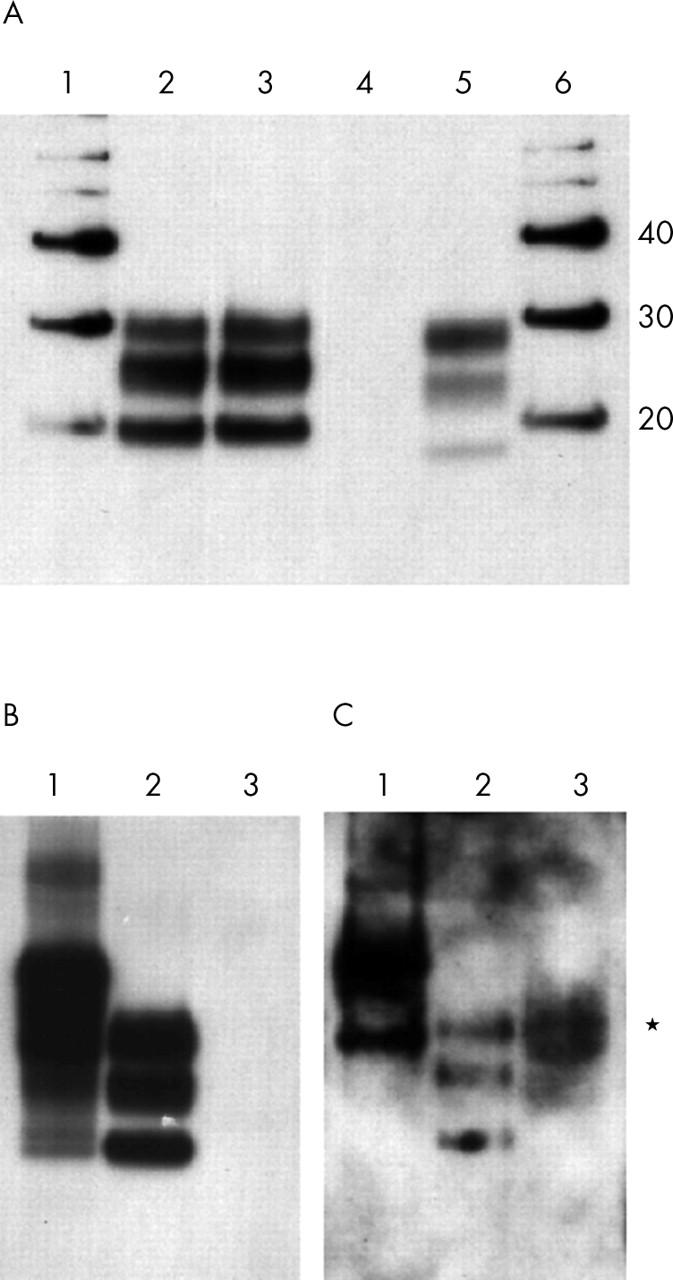
Western blot analysis of protease resistant prion protein in the retina from a patient with sporadic CJD. (A) Analysis of brain (lane 3) and retina (lane 4) from the patient, flanked by samples of a standard sporadic CJD brain (lane 2) and a standard sample of variant CJD brain (lane 5). Molecular weight markers are shown in lanes 1 and 6 with their molecular weights indicated in kDa. (B) Analysis of a second sample of retina from the patient (lane 3), alongside a standard sample of variant CJD brain before (lane 1) and after (lane 2) treatment with proteinase K. (C) 500 μl of a 10% (w/v) retinal extract from this patient is shown after precipitation with phosphotungstic acid (lane 3) and the position of the positive signal is marked (*). Lanes 1 and 2 show 1 μl of a 10% (w/v) standard homogenate of variant CJD brain before (lane 1) and after (lane 2) digestion with proteinase K. All blots were probed with the antibody 3F4.
The paraffin embedded tissue blot method was then used to assess the regional distribution of PrPSc in the fixed eye. Positive staining was seen in some (but not all) preparations of the retina. Where present, this was in the form of a bi-layer when viewed at low magnification (fig 2A). Immunohistochemistry showed that this pattern corresponded to the plexiform layers of the neural retina in which focal granular PrP staining is seen (fig 2B). The intensity of immunohistochemical staining for PrPSc was less overall than that seen in the vCJD eye, and it diminished towards the more anterior parts of the neural retina. No obvious relationship was identified between the distribution of PrPSc and other anatomical structures in the central retina. Positive immunostaining for PrPSc was not evident in the optic nerve or any ocular tissues other than neural retina by the paraffin embedded tissue blot method or by immunohistochemistry.
Figure 2.
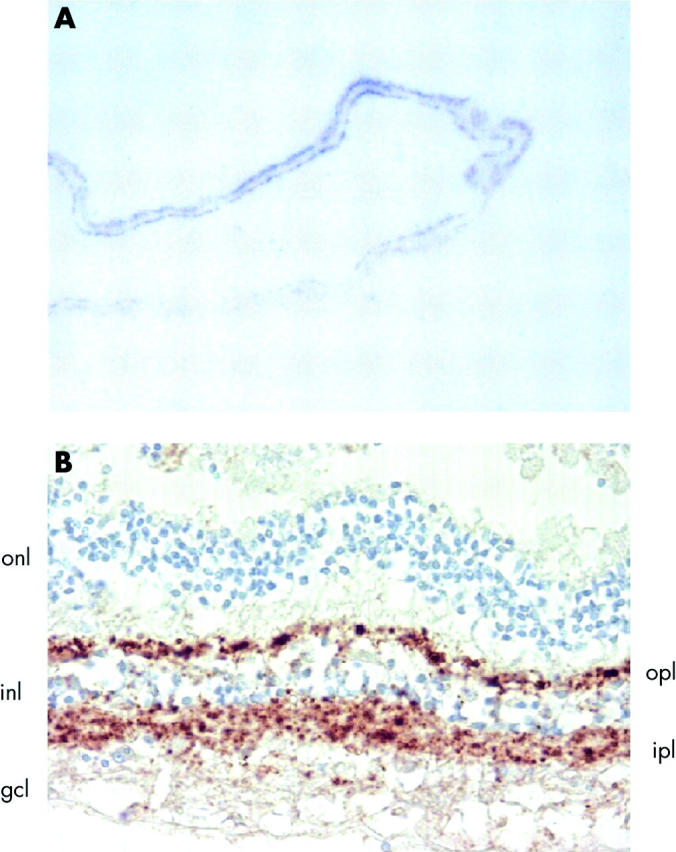
Localisation of PrPSc in the retina from a patient with sporadic CJD by (A) paraffin embedded tissue blotting with the antibody 3F4 and (B) immunohistochemistry with the antibody 12F10. The retinal layers are indicated: outer nuclear layer (onl), outer plexiform layer (opl), inner nuclear layer (inl), inner plexiform layer (ipl), ganglion cell layer (gcl).
DISCUSSION
The results presented here go some way to resolving the discrepancy between the two published studies of ocular involvement in cases of sCJD.6,8 Prominent and uniform retinal involvement appears to be a characteristic feature of vCJD and was also evident in the one case of sCJD of the VV2 subtype examined.6,8 In contrast, PrPSc is much less readily detectable in the retina in the more common MM1 subtype of sCJD. Although undetectable by biochemical assays in the MM1 sCJD case analysed by Wadsworth and colleagues, PrPSc is detectable in the retina of the MM1 sCJD case presented here, both by biochemical and histological based assays, but its level is very much lower and its distribution is perhaps less uniform. The amount of PrPSc detected in the retina by phosphotungstate precipitation assay was between one hundredth and one thousandth of the level found in the vCJD positive control brain sample.
Sporadic CJD is thought to originate from a chance conversion of the normal form of the prion protein to the abnormal form (PrPSc) in the brain in affected individuals. The presence of PrPSc outside the brain has been interpreted as centrifugal spread from the brain to the periphery, in the case of the eye, via the optic nerve with subsequent accumulation being a characteristic of regions of high synaptic density such as the plexiform layers of the retina. PrPSc positivity is known from animal studies to progress at a slow but defined rate (∼1 mm/day) along neural pathways.12 It follows that ocular involvement could be considered to be a function of the presence of PrPSc in the relevant region of the brain, the time during which it can spread via the optic nerve to the retina, and the opportunity that it has to accumulate in the synaptic layers. The mean duration of illness in sCJD is shorter than that of vCJD, and this is particularly true of the duration of the MM1 subtype of sCJD which progresses very rapidly.9,13 The cases of sCJD that we have reported are typical of their subtypes. The duration of the illness in the VV2 sCJD case we reported previously was 9 months, whereas the MM1 patient reported here had a duration of illness of only 1 month. We would predict that any larger study of ocular involvement in CJD might find a correlation between the level and extent of PrPSc accumulation in the retina and disease duration. A similar argument has recently been advanced to explain why some (but not all) cases of sCJD have detectable PrPSc in spleen and skeletal muscle.10
Irrespective of whether this general explanation is found to be correct, this study clearly indicates that the retina should be considered a potential source of transmissible infection when ophthalmic surgery is performed on any patient suffering from neurological symptoms consistent with a diagnosis of CJD. The results also underscore the importance of localisation studies in addition to sensitive biochemical tests when analysing tissues that might be heterogeneous in their accumulation of PrPSc.
Acknowledgments
The authors thank the patients’ relatives for their consent to retain and perform these studies on tissue specimens.
Abbreviations
sCJD, sporadic Creutzfeldt-Jakob disease
vCJD, variant Creutzfeldt-Jakob disease
The National CJD Surveillance Unit is funded by the UK Department of Health and the Scottish Executive and is a member of the EU TSELAB (EC QLK-CT-2002-81523) and NeuroPrion (EC FOOD-CT-2004-506579) projects.
Competing interests: none declared
Ethical approval for the acquisition and use of autopsy material for research on transmissible spongiform encephalopathies in the National CJD Surveillance Unit brain bank is covered by LREC 2000/4/157 (Professor J W Ironside).
REFERENCES
- 1.Brown P , Preece M, Brandel J-P, et al. Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 2000;55:1075–81. [DOI] [PubMed] [Google Scholar]
- 2.Llewelyn CA, Hewitt PE, Knight RSG, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 2004;363:417–21. [DOI] [PubMed] [Google Scholar]
- 3.Peden AH, Head MW, Ritchie D, et al. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004;364:527–9. [DOI] [PubMed] [Google Scholar]
- 4.S-Juan P , Ward HJT, De Silva R, et al. Ophthalmic surgery and Creutzfeldt-Jakob disease. Br J Ophthalmol 2004;88:446–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown P , Gibbs CJ, Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol 1994;35:513–29. [DOI] [PubMed] [Google Scholar]
- 6.Wadsworth JDF, Joiner S, Hill AF, et al. Tissue distribution of protease resistant prion protein in variant Creutzfeldt-Jakob disease using a highly sensitive immunoblotting assay. Lancet 2001;358:171–80. [DOI] [PubMed] [Google Scholar]
- 7.Head MW, Ritchie D, Smith N, et al. Peripheral tissue involvement in sporadic, iatrogenic, and variant Creutzfeldt-Jakob disease. Am J Pathol 2004;164:143–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Head MW, Northcott V, Rennison K, et al. Prion protein accumulation in eyes of patients with sporadic and variant Creutzfeldt-Jakob disease. Invest Ophthalmol Vis Sci 2003;44:342–6. [DOI] [PubMed] [Google Scholar]
- 9.Parchi P , Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224–33. [PubMed] [Google Scholar]
- 10.Glatzel M , Abela E, Maissen M, et al. Extraneural pathologic prion protein in sporadic Creutzfeldt-Jakob disease. N Engl J Med 2004;350:732–3. [DOI] [PubMed] [Google Scholar]
- 11.Ritchie D , Head MW, Ironside J. Advances in the detection of prion protein in peripheral tissues of variant Creutzfeldt-Jakob disease patients using paraffin-embeded tissue blotting. Neuropathol Applied Neurobiol 2004;30:360–8. [DOI] [PubMed] [Google Scholar]
- 12.Beekes M , Baldauf E, Diringer H. Sequential appearance and accumulation of pathognomonic markers in the nervous system of hamsters orally infected with scrapie. J Gen Virol 1996;77:1925–34. [DOI] [PubMed] [Google Scholar]
- 13.Pocchiari M , Puopolo M, Croes EA, et al. Predictors of survival in sporadic Creutzfeldt-Jakob disease and other human transmissible spongiform encephalopathies. Brain 2004;127:2348–59. [DOI] [PubMed] [Google Scholar]