

Abstract
Aim: To identify novel or rare rhodopsin gene mutations in patients with autosomal dominant retinitis pigmentosa and description of their clinical phenotype.
Methods: The complete rhodopsin gene was screened for mutations by DNA sequencing in index patients. Mutation specific assays were used for segregation analysis and screening for controls. Eight patients from five families and their relatives were diagnosed with autosomal dominant retinitis pigmentosa (adRP) by means of clinical evaluation.
Results: Mutation screening identified five different rhodopsin mutations including three novel mutations: Ser176Phe, Arg314fs16, and Val20Gly and two missense mutations, Pro215Leu and Thr289Pro, that were only reported once in a mutation report. Electrophysiological and psychophysical testings provide evidence of an impaired rod system with additionally affected cone system in subjects from each genotype group. Visual function tended to be less affected in subjects with the Arg314fs16 and Val20Gly mutations than in the Ser176Phe phenotype. In contrast, Pro215Leu and Thr289Pro mutations caused a remarkably severe phenotype.
Conclusion: The ophthalmic findings support a correlation between disease expression and structural alteration: (1) extracellular/intradiscal Val20Gly and cytoplasmic Arg314fs16 mutation—mild adRP phenotype; (2) Ser176Phe mutation—“mostly type 1” disease; (3) predicted alteration of transmembrane domains TM V and TM VII induced by Pro215Leu and Thr289Pro—severe phenotype. However, variation of phenotype expression in identical genotypes may still be a typical feature of RHO mutations.
Keywords: retinitis pigmentosa, rhodopsin, phenotype, mutation, autosomal dominant
Retinitis pigmentosa (RP) is a clinically and genetically heterogeneous group of inherited retinal degenerations.1 Patients with RP experience night blindness, visual field constriction, and eventually loss of central vision, in most cases caused by degeneration of the photoreceptor cells of the retina.1,2 There are autosomal dominant (adRP), autosomal recessive (arRP), X linked (xlRP), and rare mitochondrial or digenic forms.1 To date, 13 gene loci are known for adRP,3,4,5,6,7,8,9,10,11,12,13 21 for arRP, and five for xlRP (RetNet http://www.sph.uth.tmc.edu/RetNet/).
The first photoreceptor specific gene found to be mutated in adRP was rhodopsin (RHO).14,15 Rhodopsin is the light absorbing molecule that initiates the signal transduction cascade in rod photoreceptors. Mutations of the rhodopsin gene account for approximately 25% of adRP.16 Numerous mutations in the RHO gene have been identified, most of them being point mutations.15 The analysis of mutant rhodopsins suggest that some RP mutations impair protein folding, 11-cis retinal chromophore binding, G-protein coupling/activation, and/or cellular trafficking of the rhodopsin protein.17,18
In this study we report the results of the RHO gene screening in five independent adRP families.
PATIENTS AND METHODS
Recruitment of patients, DNA isolation, and mutation analysis
Eight patients were included into the study, all suffering from adRP. The study was conducted in accordance with the tenets of the Declaration of Helsinki.
Mutation detection by direct sequencing
Patient DNA was extracted using a standard salting out procedure.19 For mutation detection by sequencing, PCR was performed using corresponding sense and antisense primers and 1 U Taq polymerase (Eppendorf, Hamburg, Germany). PCR products were separated on a DNA capillary sequencer (ABI 3100 Genetic Analyzer, Applied Biosystems, Foster City, CA, USA).
Mutation detection by DHPLC
Denaturing high performance liquid chromatography (DHPLC) analysis was conducted with the WAVE nucleic acid fragment analysis system equipped with a L-7400 UV detector (Transgenomic, Omaha, NE, USA). Samples with aberrant profiles were sequenced.
Mutation detection by RFLP
A 588 bp PCR product encompassing Exon 1 was digested with 1 U RsaI restriction enzyme (NEB, Beverly, MA, USA). The Val20Gly missense change results in the loss of one of three genuine RsaI restriction sites. Restriction digest for normal individuals results in two fragments of about 420 bp and 160 bp that can be visualised on a 4% agarose gel. For individuals heterozygous for the Val20Gly mutation, restriction digest results in an additional fragment of 178 bp.
Clinical studies of the RHO mutation phenotype
Phenotype analysis comprised clinical examination, Goldmann perimetry, Panel D15 testing, dark adapted final thresholds, Ganzfeld electroretinography (UTAS 2000 system; LKC Technologies, Gaithersburg, USA), according to ISCEV standard20 and multifocal electroretinography using the VERIS system (EDI, San Francisco, CA, USA).21,22
RESULTS
Mutation analysis
The index patient of each family was screened for mutations in the RHO gene. Table 1 summarises the respective sequence alterations. Three of them were novel mutations (Val20Gly, Ser176Phe, Arg314fs16), and the other two mutations (Pro215Leu, Thr289Pro) have only been reported once in a brief mutation report without clinical details.23
Table 1.
RHO mutations
| Nucleotide sequence alteration | Consequence | Location | Mutation carriers/families tested | Mutation carriers/controls | Previously reported |
| c.59T>G | Val20Gly | Exon 1 | 6/10 | 0/100 | No |
| c.527C>T | Ser176Phe | Exon 2 | 1/1 | 0/100 | No |
| c.644C>T | Pro215Leu | Exon 3 | 5/12 | 0/100 | See ref 23 |
| c.865°>C | Thr289Pro | Exon 4 | 1/1 | 0/100 | See ref 23 |
| c.942insG | Arg314fs16 | Exon 5 | 1/1 | Not performed | No |
Clinical studies
Figure 1 (A to E) shows the pedigree of the patients/families investigated.
Figure 1.
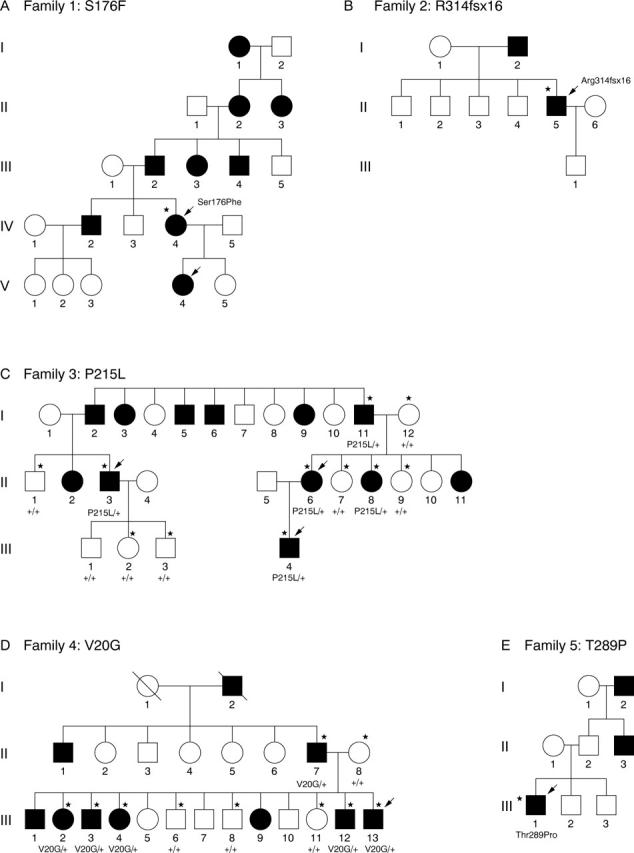
(A to E) Pedigrees of adRP families. Circles, females; squares, males; solid symbols, affected members; open symbols, unaffected members; slashed symbols, deceased members; arrows, persons examined ophthalmologically; asterisks, DNA analysis performed.
The clinical characteristics of affected patients are summarised in table 2. Original phenotype data is indicated in figures 2–4.
Table 2.
RHO Mutations and Phenotype
| Family number/mutation | Age of onset/mode of onset (age) | VA: RE/LE initial presentation (age) | Refractive error: RE/LE | Visual field: RE/LE (age) | ERG | Fundus | DA/panel D15 |
| 1/Ser176Phe (German) | IV:4 Night blindness; field constriction (13) | 60/100; 60/100 (33) | −1.75/−1.75 | Constriction to 15° (III/4e 90°) (33) | Scotopic: noise level | Slight optic atrophy, vessel narrowing, absent macular reflexes, peripheral pigment mottling, slight peripheral hyperpigmentation | Significant elevation/desaturated: normal |
| Loss visual acuity (30) | Cone 30Hz-flicker: residual responses (33) | ||||||
| V:4 Night blindness, glare sensitivity (2) | 80/100; 100/100 (6) | +1.5/+1.5 | Near normal (III/4e), constriction to 10–20° (I/4e 90°) (6) | not performed | Normal optic discs, peripheral RPE atrophy without bone spicules | Desaturated: normal | |
| 2/Arg314fs16 (Bosnian) | II:5 Night blindness, glare sensitivity, field constriction (33) | 100/100; 120/100 (36) | Emmetropic | Constriction to 50–60° with large scotomas 15–40° (III/4e 90°) (36) | Scotopic: amplitude reduction to 10% normal range | Mild optic atrophy, moderately narrowed vessels, atypical macular ILM reflexes; bone-spicules in lower sector, diffuse RPE atrophy | Significant elevation/desaturated: normal |
| Photopic: amplitude reduction to 50% normal range | |||||||
| MfERG: preserved foveal cone responses | |||||||
| 3/Pro215Leu (German) | II:3 Night blindness (birth), visual acuity loss, field constriction (33), glare sensitivity (45) | 10/200; 10/200 (48) | +1,25/+1,25 | Constriction to 5–7° (III/4e 90°) (48) | Noise level (48) | Waxy optic nerve atrophy, vessel attenuation, RPE atrophy, and hyperpigmentation | Significant elevation/saturated: disturbances all axes |
| II:6 Night blindness (birth) | No details | No details | Constriction to 5–10° (III/4e 90°) (35) | Noise level (35) | No details | No details | |
| III:4 Night blindness (birth), not aware of other symptoms | 120/100; 80/100 (16) | +2.5/+2.75 | Constriction to 10–25° (right eye), 7–15°(left eye) (III/4e 90°) (16) | Rod responses: noise level; cone 30 Hz: residual responses (16) | Normal optic disc, slightly narrowed vessels, cystoid macula oedema, peripheral RPE atrophy with hyperpigmentation | Significant elevation/desaturated: slight disturbances all axes | |
| 4/Val20Gly (Austrian) | III:13 No symptoms | 100/100; 80/100 (34) | 0; +3.5 RE: cataract surgery at age 27 | Not performed | Scotopic and photopic amplitude reduction with rest function; Electro-oculogram: no light peak, pathological Arden ratio | Absent macular reflexes, narrowed vessels, mid-peripheral hyperpigmentation | No details |
| 5/Thr289Pro (Austrian) | III:1 Night blindness, field constriction (3), color vision (9), glare sensitivity (12), visual acuity (18), cataract extraction (27) | 10/250; 10/250 (27) | Emmetropic | Constriction to <5° (III/4e 90°) | Scotopic/photopic noise level MfERG noise level | Temporal optic disc pallor, no macular reflexes, mid-peripheral hyperpigmentation | Significant elevation |
Figure 2.
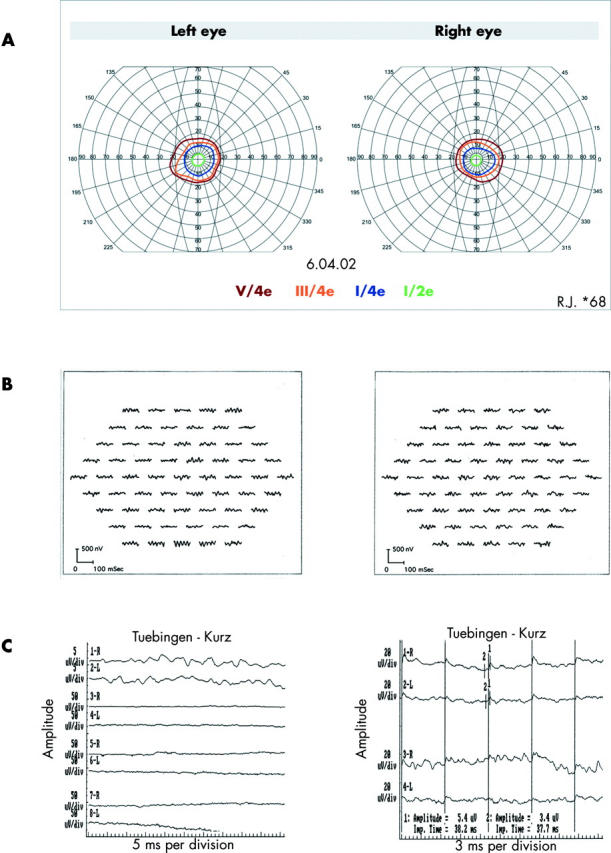
Patient IV:4 (family 1; Ser176Phe). (A) Perimetry (targets V/4e, III/4e, I/4e, and I/2e). (B) MfERG responses showing 61 single traces of right eye (RE) (right lane) and left eye (LE) (left lane). (C) Ganzfeld-ERG. Left column: waveforms (top to bottom) are isolated rod b-wave of RE (1-R, 3-R, 5-R) and LE (2-L, 4-L, 6-L), scotopic mixed rod/cone response of RE (7-R) and LE (8-L). Right column: photopic 30 Hz flicker response of RE (1-R) and LE (2-L). Traces 3-R and 4-L show 30 Hz repetition. “1”: peaks of the 30 Hz flicker response. Left column: calibrations: 5 microV/div;5 ms/div (traces 1-R; 2-L) and 50 microV/div;5 ms/div for other traces. Right column: calibrations 20 microV/div;3 ms/div.
Figure 4.

(A) Fundus LE (patient II:3; family 3; Pro215Leu). Nasal periphery (top) and posterior pole (bottom). (B) Fundus LE (patient III:4; family 3; Pro215Leu). Nasal periphery (top) and posterior pole (bottom).
DISCUSSION
We studied patients from five independent families showing typical clinical features of autosomal dominant RP. Two of the novel mutations were missense mutations, and one was a 1bp insertion (c.942insG) that results in a frameshift and subsequent translation termination.
We found considerable relation between the individual mutation and disease expression. Cideciyan and colleagues distinguished two patterns of rod disease expression in a variety of rhodopsin mutations.24 Other widely accepted classification systems have been developed by Massof & Finkelstein,25 Lyness et al,26 and Fishman et al.27 According to the latter systems the phenotypes of our study can be subdivided into distinct groups.
Patients with the novel mutations Arg314fs16 (family 2) and Val20Gly (family 4) express a remarkably mild “mostly type 2” phenotype25 with late onset of symptoms and a more favourable visual prognosis or “R”-type26 and the Ser176Phe phenotype (family 1) discloses an intermediate “mostly type 1” or “D-” phenotype. In contrast, the Pro215Leu (family 3) and Thr289Pro (family 5) mutations result in a severe type 1- or D-phenotype with early onset of symptoms and rapid loss of visual field area, corresponding with a diffuse and progressive loss of rod and cone function.
Based on current models of rhodopsin (fig 5), two of the novel mutations (Val20Gly and Ser176Phe) involve amino acids on the intradiscal/extracellular side and one occurs at the cytoplasmic side (Arg314fs16). Mutations Pro215Leu and Thr289Pro involve transmembrane domains.
Figure 5.

Two dimensional model of rhodopsin (mutations boxed). The TM helices are labelled I-VII. Arg314fs16 mutation: Arg-Glu-Leu-His-Ala-His-His-His-Leu-Leu-Arg-Gln-Glu-Pro-Thr-Gly-STOP.
Considering intradiscal/extracellular mutations, it has been reasoned that missense mutations affecting residues 2–428 or 15–1729 interfere with N-glycosylation or that the replacement of cysteines 110 and 187 prevent the formation of the disulfide bridge.15
It could be speculated that the Ser176Phe mutation may induce a structural alteration of the cysteine 187 neighbourhood.
The Val20Gly missense mutation is close to previously described mutations in the N-terminal region of the polypeptide. The phenotype of patient III:13 carrying this novel mutation was extremely mild with moderately affected rod and cone function even at age 34.
The c.942insG mutation causes a frameshift mutation with premature stop codon that results in an alteration and shortening of the cytoplasmatic domain, the first rhodopsin mutation to be described in a Bosnian family. The phenotype of the single available patient is relatively mild with late onset symptoms and relatively well preserved central cone function. Cideciyan and colleagues described the phenotype of other C-terminal truncated mutations (Gln312X and Gln344X) and classified them as class B mutants.24
The Pro215Leu and Thr289Pro mutations involve amino acid exchanges within transmembrane domains, and both mutations eliminate or introduce proline residues. Recent publications described mutations perturbing critical interhelical interactions between TM III and TM V, namely the Glu122, His211 salt bridge, resulting in a severe type of adRP in vivo.30–33
The Thr289Pro phenotype (family 5, individual III:1) may serve as another example of severe type adRP induced by a missense mutation in TMVII. Within TM VII, 11-cis-retinal covalently binds to opsin at the epsilon amino group of Lys296.34,35 The phenotype presented in our study parallels many features of the unusually severe Lys296Glu phenotype.36,37
Both intrafamilial and interfamilial phenotype differences among carriers of identical rhodopsin mutations have been described.38,39 On the other hand, a disease course fairly constant in all affected persons within the same large pedigree has also been documented.40
In summary, the phenotype description of Ser176Phe gives at least consistent examples of mother (IV:4) and daughter (V:4) expressing an intermediate type of adRP with early onset of symptoms, but possibly long time preserved visual acuity. For the Arg314fsX16 and Val20Gly mutations, we could describe an unusually mild phenotype. The alteration of transmembrane helices TM V or TM VII by the Pro215Leu and Thr289Pro missense mutation lead to severe adRP. However, more interfamilial and intrafamilial clinical data will be necessary to draw conclusions on a constant genotype-phenotype correlation in these mutations.
Figure 3.
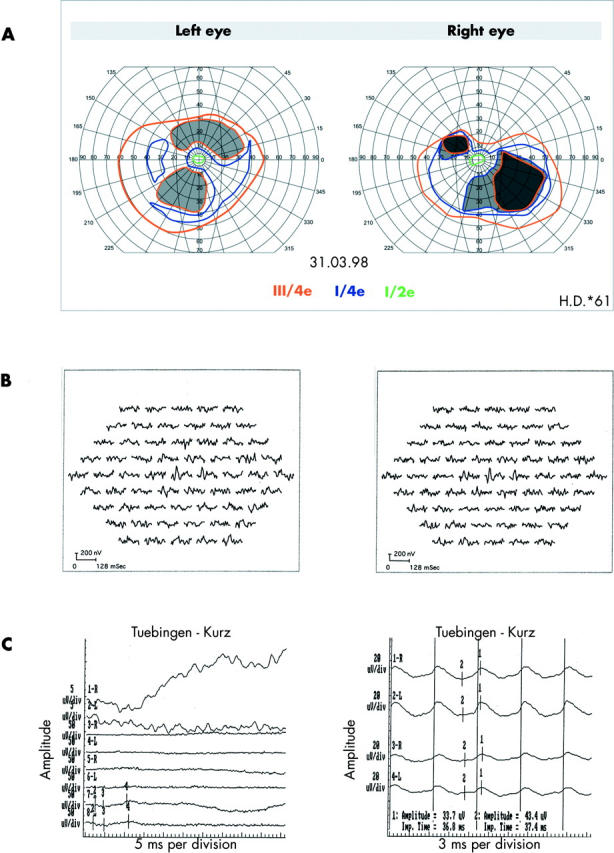
Data of patient II:5 (family 2; Arg314fs16). (A) Perimetry (targets III/4e, I/4e, and I/2e). (B) See figure 2B. (C) Ganzfeld-ERG: waveforms are indicated as in figure 2. “3” indicates a-wave, “4” indicates b-wave peak. Peaks of 30 Hz flicker signals are indicated by “1”. Calibrations: see figure 2.
Acknowledgments
We are grateful to Dr Daxecker and Dr Lalehabbasi, Innsbruck/Austria for collaboration
Abbreviations
adRP, autosomal dominant retinitis pigmentosa
arRP, autosomal recessive retinitis pigmentosa
xlRP, X linked retinitis pigmentosa
DHPLC, denaturing high performance liquid chromatography
Supported by the grants ZR 1/16-1 and ZR 1/16-2 of the Deutsche Forschungsgemeinschaft (DFG)
REFERENCES
- 1.Heckenlively JR, Daiger SP. In: Rimon DL, Conner JM, Pyeritz RE, eds. Hereditary retinal and choroidal degenerations. New York: Churchill Livingstone, 2001:2255–576.
- 2.Bird AC. Retinal photoreceptor dystrophies. Am J Ophthalmol 1995;19:543–62. [DOI] [PubMed] [Google Scholar]
- 3.Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990;343:364–6. [DOI] [PubMed] [Google Scholar]
- 4.Kajiwara K, Hahn LB, Mukai S, et al. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature 1991;354:480–3. [DOI] [PubMed] [Google Scholar]
- 5.Farrar GJ, Kenna P, Jordan SA, et al. A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature 1991;354:478–80. [DOI] [PubMed] [Google Scholar]
- 6.Bessant DA, Payne AM, Mitton KP, et al. A mutation in NRL is associated with autosomal dominant retinitis pigmentosa. Nat Genet 1999;21:355–6. [DOI] [PubMed] [Google Scholar]
- 7.Sullivan LS, Heckenlively JR, Bowne SJ, et al. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet 1999;22:255–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sohocki MM, Sullivan LS, Mintz-Hittner HA, et al. A range of clinical phenotypes associated with mutations in CRX, a photoreceptor transcription-factor gene. Am J Hum Genet 1998;63:1307–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wada Y, Abe T, Takeshita T, et al. Mutation of human retinal fascin gene (FSCN2) causes autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 2001;42:2395–400. [PubMed] [Google Scholar]
- 10.Bowne SJ, Sullivan LS, Blanton SH, et al. Mutations in the inosine monophosphate dehydrogenase 1 gene (IMPDH1) cause the RP10 form of autosomal dominant retinitis pigmentosa. Hum Mol Genet 2002;11:559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kennan A, Aherne A, Palfi A, et al. Identification of an IMPDH1 mutation in autosomal dominant retinitis pigmentosa (RP10) revealed following comparative microarray analysis of transcripts derived from retinas of wild-type and Rho(_/_) mice. Hum Mol Genet 2002;11:547–57. [DOI] [PubMed] [Google Scholar]
- 12.Keen TJ, Hims MM, McKie AB, et al. Mutations in a protein target of the Pim-1 kinase associated with the RP9 form of autosomal dominant retinitis pigmentosa. Eur J Hum Genet 2002;10:245–9. [DOI] [PubMed] [Google Scholar]
- 13.Rebello G, Ramesar R, Vorster A, et al. Apoptosis-inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci USA 2004;101:6617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dryja TP, Li T. Molecular genetics of retinitis pigmentosa. Hum Mol Genet 1995;4:1739–43. [DOI] [PubMed] [Google Scholar]
- 15.Gal A, Apfelstedt-Sylla E, Janecke AR, et al. Rhodopsin mutations in inherited retinal dystrophies and dysfunctions. Prog Retinal Eye Res 1997;16:51–79. [Google Scholar]
- 16.Chuang J-Z, Vega C, Jun W, et al. Structural and functional impairment of endocytic pathways by retinitis pigmentosa mutant rhodopsin-arrestin complexes. J Clin Invest 2004;114:131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Franke RR, Sakmar TP, Graham RM, et al. Structure and function in rhodopsin. Studies of the interaction between the rhodopsin cytoplasmic domain and transducin. J Biol Chem 1992;267:14767–74. [PubMed] [Google Scholar]
- 18.Sung CH, Schneider BG, Agarwal N, et al. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A 1991a;88:8840–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1988;16:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marmor MF, Zrenner E. Standard for clinical electroretinography (1999 update). Doc Ophthalmol 1999;97:143–56. [DOI] [PubMed] [Google Scholar]
- 21.Sutter EE, Tran E. The field topography of ERG components in man, I: the photopic luminance response. Vis Res 1992;32:433–66. [DOI] [PubMed] [Google Scholar]
- 22.Kretschmann U, Seeliger MW, Rüther K, et al. Multifocal electroretinography in patients with Stargardt’s macular dystrophy. Br J Ophthalmol 1998;82:267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Gimeno M, Trujillo MJ, Lorda I, et al. Three novel mutations (P215L, T289P, and 3811-2 A-->G) in the rhodopsin gene in autosomal dominant retinitis pigmentosa in Spanish families. Hum Mutat 2000;16:95–6. [DOI] [PubMed] [Google Scholar]
- 24.Cideciyan AV, Hood DC, Huang Y, et al. Disease sequence from mutant rhodopsin allele to rod and cone photoreceptor degeneration in man. Proc Natl Acad Sci U S A 1998;95:7103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Massof RW, Finkelstein D. Two forms of autosomal dominant primary retinitis pigmentosa. Doc Ophthalmol 1981;51:289–346. [DOI] [PubMed] [Google Scholar]
- 26.Lyness AL, Ernst W, Quinlan MP, et al. A clinical, psychophysical, and electroretinographic survey of patients with autosomal dominant retinitis pigmentosa. Br J Ophthalmol 1985;69:326–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fishman GA, Alexander KR, Anderson RJ. Autosomal dominant retinitis pigmentosa. A method of classification. Arch Ophthalmol 1985;103:366–74. [DOI] [PubMed] [Google Scholar]
- 28.Bunge S, Wedemann H, David D, et al. Molecular analysis and genetic mapping of the rhodopsin gene in families with autosomal dominant retinitis pigmentosa. Genomics 1993;17:230–3. [DOI] [PubMed] [Google Scholar]
- 29.Sung CH, Davenport CM, Hennessey JC, et al. Rhodopsin mutations in autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A 1991b;88:6481–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel AB, Crocker E, Eilers M, et al. Coupling of retinal isomerization to the activation of rhodopsin. Proc Natl Acad Sci U S A 2004;101:10048–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stojanovic A, Hwang I, Khorana HG, et al. Retinitis pigmentosa rhodopsin mutations L125R and A164V perturb critical interhelical interactions: new insights through compensatory mutations and crystal structure analysis. J Biol Chem 2003;278:39020–8. [DOI] [PubMed] [Google Scholar]
- 32.Dryja TP, Hahn LB, Cowley GS, et al. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci U S A 1991;88:9370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs S, Kranich H, Denton MJ, et al. Three novel rhodopsin mutations (C110F, L131P, A164V) in patients with autosomal dominant retinitis pigmentosa. Hum Mol Genet 1994;3:1203. [DOI] [PubMed] [Google Scholar]
- 34.Bownds D. Site of attachment of retinal in rhodopsin. Nature 1967;216:1178–81. [DOI] [PubMed] [Google Scholar]
- 35.Wang JK, McDowell JH, Hargrave PA. Site of attachment of 11-cis-retinal in bovine rhodopsin. Biochemistry 1980;19:5111–17. [DOI] [PubMed] [Google Scholar]
- 36.Keen TJ, Inglehearn CF, Lester DH, et al. Autosomal dominant retinitis pigmentosa: four new mutations in rhodopsin, one of them in the retinal attachment site. Genomics 1991;11:199–205. [DOI] [PubMed] [Google Scholar]
- 37.Owens SL, Fitzke FW, Inglehearn CF, et al. Ocular manifestations in autosomal dominant retinitis pigmentosa with a Lys-296-Glu rhodopsin mutation at the retinal binding site. Br J Ophthalmol 1994;78:353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berson EL, Rosner B, Sandberg MA, et al. Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro23His). Arch Ophthalmol 1991a;109:92–101. [DOI] [PubMed] [Google Scholar]
- 39.Berson EL, Rosner B, Sandberg MA, et al. Ocular findings in patients with autosomal dominant retinitis pigmentosa with rhodopsin, proline-347-leucine. Am J Ophthalmol 1991b;111:614–23. [DOI] [PubMed] [Google Scholar]
- 40.Rosas DJ, Roman AJ, Weissbrod P, et al. Autosomal dominant retinitis pigmentosa in a large family: a clinical and molecular genetic study. Invest Ophthalmol Vis Sci 1994;35:3134–44. [PubMed] [Google Scholar]