

Abstract
Background/aims: Neuronal degeneration has been reported to occur in diabetic retinopathy before the onset of detectable microvascular abnormalities. To investigate whether advanced glycation end products (AGE) could be directly responsible for retinal neurodegeneration, retinal explants were incubated with glycated bovine serum albumin (BSA).
Methods: Retinal explants obtained from non-diabetic adult rats were incubated 4 days with or without 200 μg/ml glycated BSA. Neural apoptosis was quantified by terminal dUTP nick end labelling (TUNEL) binding and immunostaining with anti-cleaved caspase-3 antibody. Expression of glial fibrillary acidic protein (GFAP) was localised by immunofluorescence.
Results: TUNEL and cleaved caspase-3 positive cells increased significantly by 2.2-fold and 2.5-fold in retinal explants incubated in glycated BSA (p<0.05), respectively. The ganglion cell layer was the most sensitive retinal layer to the glycated BSA. Neuronal degeneration was confirmed by the increased GFAP labelling in Müller glial cells from retinal explants treated with glycated BSA.
Conclusion: These results suggest that AGE could induce retinal neurodegeneration in the absence of blood perfusion. Cells in the ganglion cell layer appeared to be the most sensitive as in diabetic retinopathy and its animal models. AGE toxicity could therefore contribute to the early pathological mechanisms of diabetic retinopathy.
Keywords: advanced glycation end products, diabetic retinopathy, glial reaction, neuronal apoptosis, rat
Neural cell depletion was observed decades ago in diabetic patients.1 Although diabetic retinopathy (DR) is classically considered a retinal microangiopathy, loss of colour vision, of contrast sensitivity, electroretinogram abnormalities, and neuronal apoptosis were detected in diabetic patients before capillary closure and microaneurysms, suggesting that DR could have a neuropathic component that may occur early in the disease.2,3 These observations have been confirmed by animal studies showing that neuronal apoptosis and glial reaction could occur after only 1 month of experimental diabetes in streptozotocin induced diabetic rat.3,4
The mechanisms by which diabetes induces neuroglial reactions remain unknown. These neuroglial reactions are unlikely to be the result of ischaemic necrosis since they occur very early in diabetic rats before the onset of any known vascular occlusion.3,4 However, neural cell death due to increased vascular permeability cannot be ruled out, since some increase in permeability can occur after 2–4 weeks of experimental diabetes in rats.3,4 The advanced glycated end products (AGE) can provide the early molecular pathogenic mechanisms responsible for neuronal apoptosis and neuroglial reactions. Their involvement in the development of DR is supported by the prevention of DR by blockers of AGE formation in animal models.5,6 The potential retinal toxicity of AGE was consistent with apoptosis of cultured cortical neurons7 and with their accumulation in the retina of diabetic patients.8
The present study was performed to assess whether AGE can directly trigger neuronal apoptosis in the retina, independently of blood vessel circulation. AGE were applied to retinal explants, as they both preserve the retinal cellular architecture and limit the influence of the blood vasculature.
MATERIALS AND METHODS
Male non-diabetic Long Evans rats (Charles River Laboratories, France) aged 7 weeks were used for this study. Animal studies conformed to the principles of laboratory animals (NIH publication no 85–23, revised 1985), and the French law on animal protection.
Retinal explants were obtained according to the protocol described by Pinzon-Duarte et al.9 The choroid was gently peeled away from the retinal pigment epithelium (RPE), leaving the RPE attached to the neurosensory retina. The retina was transferred onto a polycarbonate membrane (Transwell, Corning, Netherlands) and cultured in Dulbecco’s Modified Eagle Medium containing 10% fetal calf serum and 10 μg/ml gentamicin. For each animal, one retina was used as a control retinal explant and the other retina was incubated in 200 μg/ml glycated BSA (Sigma Chemical Co, St Louis, MO, USA, 95% purity) for 4 days, a period providing the lowest variability when inducing apoptosis in such ex vivo retina.10 This glycated BSA concentration was selected because similar concentrations (250 μg/ml; 100 μg/ml) were used previously to demonstrate AGE neurotoxicity7 and AGE role in increased retinal expression of vascular endothelium growth factor (VEGF).11 Furthermore, circulating AGE ranged in diabetic patients between 1–120 μg/ml, which was considered equivalent to 4–480 μg/ml glycated BSA by the authors of the study.12 No albumin was added to the control condition because the culture medium already contains albumin (2700 μg/ml) from the fetal calf serum such that the addition of glycated BSA represented less than 8% increase of its concentration.
The retinal tissue was fixed and processed for immunohistochemistry and terminal dUTP nick end labelling-fluorescein-isothiocyanate (TUNEL) labelling, as previously described.10,13 Retinal explant sections were labelled with anti-cleaved active caspase-3 antibody (Cell Signalling Technology, Herts, UK, 1:100), anti-vimentin antibody (Chemicon International, Temecula, CA, USA, 1:200), anti-glial fibrillary acidic protein (GFAP) antibody (Dako, Trappes, France, 1:50), anti-l-glutamate/l-aspartate transporter antibody (GLAST) (Chemicon, 1:200), and anti-glutamine synthetase antibody (Chemicon, 1:400). Nuclear labelling was achieved by incubating the sections in diamidino-phenyl-indole (DAPI) solution (Sigma, solution stock: 500 ng/ml concentration, 1:400).
Cells were counted under the 40× objective on the microscope (Olympus, Melville, NY, USA) in five visual fields (250 μm long) from the retinal explant section. The number of cells positive for the TUNEL staining or the cleaved caspase-3 immunolabelling was normalised to the DAPI labelled retinal cell nuclei. Data were statistically analysed with the Mann-Whitney rank sum test.
RESULTS
AGE induced retinal cell apoptosis
The treatment with glycated BSA for 4 days induced a 2.1-fold increase in the number of TUNEL positive cells (glycated BSA: 14.44% (SD0.62%), n = 7; control: 6.63% (0.46%), n = 7, p <0.001), which was statistically significant in all nuclear retinal layers (fig 1A, B, E). Small groups of adjacent TUNEL positive cells were furthermore observed in the inner part of the outer nuclear layer (ONL) in four retinal explants treated with glycated BSA (n = 7) (fig 1B), whereas these characteristics were not observed in control retinal explants (fig 1A).
Figure 1.
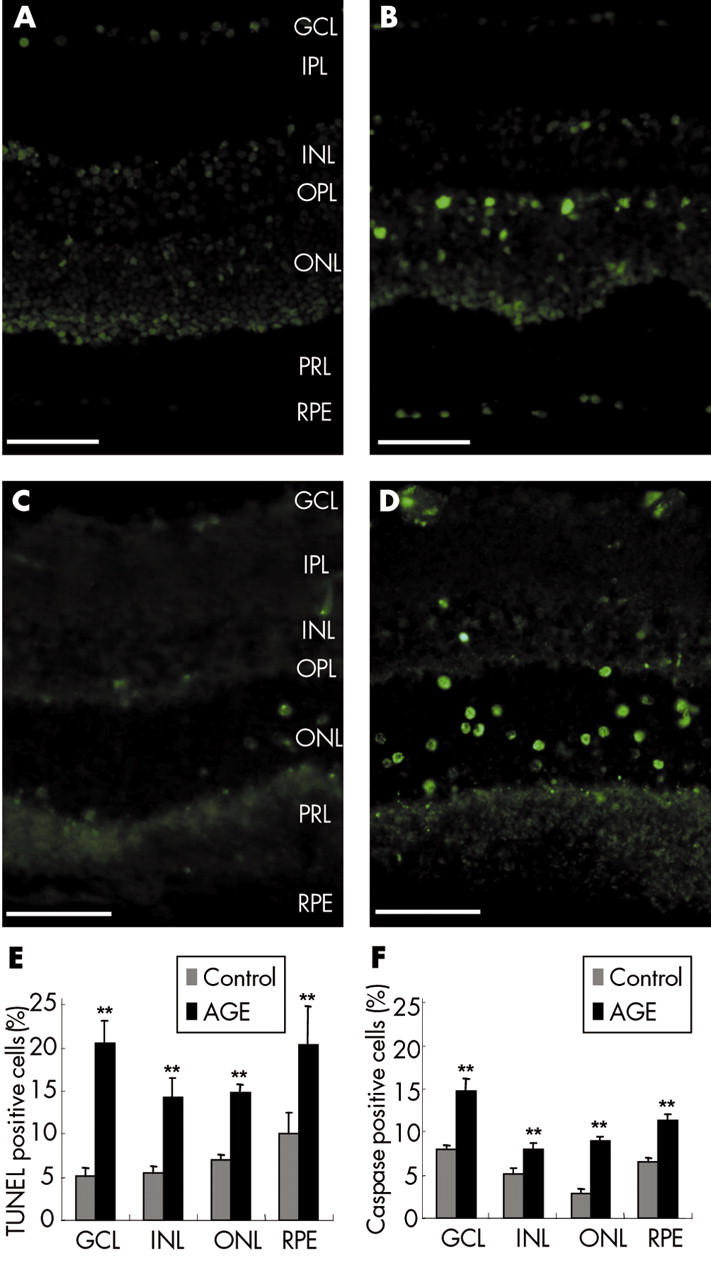
Retinal cell apoptosis induced by glycated BSA. Terminal dUTP nick end labelling (TUNEL-FITC) of cryostat sections from control retinal explants (A) or explants treated with glycated BSA (B). Immunolabelling for the active cleaved caspase-3 on cryostat sections from control retinal explants (C) and explants treated with glycated BSA (D). Note that small groups of adjacent TUNEL positive cells can be observed in the inner part of the outer nuclear layer in the retinal explant treated with glycated BSA (B) while these features are not found in the control retinal explant (A). (E) Quantification of the TUNEL positive retinal cells in control retinal explants (n = 7) and explants treated with glycated BSA (n = 7). (F) Quantification of cleaved active caspase-3 positive cells in control retinal explants (n = 7) and explants treated with glycated BSA (n = 7). In all retinal layers, the number of TUNEL and caspase-3 positive cells were significantly greater in retinal explants treated with glycated BSA than in control explants (p<0.001). GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; PRL, photoreceptor layer, RPE, retinal pigment epithelium. Errors bars indicate SEM. Scale bars represent 50 μm.
To confirm the retinal toxicity elicited by glycated BSA, retinal explant sections were labelled with an earlier marker of apoptosis, the anti-cleaved active caspase-3 antibody (fig 1C, D). The treatment with glycated BSA induced a 2.4-fold increase in active caspase-3 positive cells (glycated BSA: 8.59% (0.39%), n = 7; control: 3.48% (0.44%), n = 7, p <0.001), statistically significant in all nuclear retinal layers (fig 1F). These data suggest that a glycated protein can induce neuronal apoptosis in the retina as early as 4 days of incubation.
AGE induced glial reaction
Neuronal apoptosis is classically associated to a glial reaction which is demonstrated by a local increase in GFAP expression in the retina at the site of the lesion.13 In control retinal explants, GFAP was restricted to the innermost retinal layer, where astrocytes are located, and to the outer plexiform layer (OPL). Limited GFAP positive Müller cell processes were occasionally observed (fig 2A, C). In contrast, in retinal explants treated with glycated BSA, all Müller cell processes were extensively GFAP positive from their end tip at the inner limiting membrane to their opposite end at the outer limiting membrane. The GFAP staining was not locally restricted but extended throughout the entire retinal explant sections (fig 2B, D). No change in GLAST and glutamine synthetase was detected following the AGE treatment (data not shown). The increase in GFAP expression showed that a treatment with a glycated protein can induce a glial reaction.
Figure 2.

Glial reaction induced by the glycated BSA. GFAP immunostaining in cryostat sections of control retinal explants (A, C) and explants treated with glycated BSA (B, D). In control retinal explants (A, C), GFAP staining is localised to the innermost retinal layer, where astrocytes are located, and to the outer plexiform layer (OPL). In contrast, in retinal explants treated with glycated BSA (B, D), Müller cell processes are extensively GFAP positive from their end tips at the inner limiting membrane to their opposite end at the outer limiting membrane. This increased GFAP staining is observed on the whole retinal explant section. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; PRL, photoreceptor layer, RPE, retinal pigment epithelium. Scale bars represent 50 μm.
DISCUSSION
Electrophysiological measurements in diabetic patients and animal models generally have located the earlier deficits to the inner retina but some series have indicated visual dysfunction in the outer retina and RPE.2 Histologically, although neuronal apoptosis has been prominent in the ganglion cell layer (GCL),3 the cell loss also affected the inner nuclear layer (INL) and has been occasionally reported in the ONL.14 In our retinal explants treated with glycated BSA, neuronal apoptosis was more evenly distributed in all retinal layers, but was predominant in the GCL and ONL. These observations were consistent with those obtained after incubation of retinal cultures with the AGE precursor glyoxal.15 However, apoptotic cells accounted for only 14% in the 4 day glycated BSA incubation with a maximum of 20% in the GCL, whereas Reber et al15 reported up to 50% retinal cell death after only 9 hour glyoxal incubation with a similar higher sensitivity of the GCL. The difference may be the result of the different mechanisms involved, as glyoxal cannot only generate AGE but also reactive species, whereas in our experiment, cell toxicity can only result from the incubated glycated BSA. Our results indicated further that retinal cell death can occur by activation of caspase-3, although they do not exclude the contribution of other death pathways like caspase independent mechanisms.16
In diabetic patients and animal models, Müller glial cells are also affected as indicated by their increased GFAP expression.3,4 Under our experimental conditions, Müller glial cells also exhibited a major upregulation of GFAP expression after 4 days of incubation in glycated BSA. This Müller cell change is likely to result from the neuronal apoptosis induced by glycated BSA. However, we cannot exclude that the Müller cell change could result from a direct effect of glycated BSA on these cells. The presence of a large mass of TUNEL positive materials may further indicate that glial cells have impaired phagocytic abilities, as previously reported for resident peritoneal macrophages in streptozotocin induced diabetic animals17 or in vitro following AGE incubation.18
Neuroglial alterations are early events in the development of DR. Our study using retinal explants suggest that neuroglial lesions could result from AGE diffusion into the retinal tissue independently of major vascular perturbations like occlusions. Our study further underlines that AGE do not necessarily require to be synthesised into the retinal tissue to induce neuroglial reactions, as incubation of the retinal tissue with glycated BSA is sufficient to induce these lesions. Unless synthesised in retinal cells like Müller cells that participate in the retinal blood barrier and stock glucose,3 AGE need to cross the retinal-blood barrier in order to affect retinal cells. Breakdown of the retinal-blood barrier can be induced by AGE themselves, or by other molecules released during diabetic conditions, like VEGF, which synthesis is stimulated by AGE.6,11 The required diffusion of AGE from the circulation would suggest that an increase in permeability of the retinal-blood barrier occurs before the neuroglial reaction in diabetic retinopathy. The molecular pathways of AGE induced neuroglial reactions are not known but could be related to AGE receptors like RAGE receptors, that are located in the inner retina in humans and rats19 or the galectin-3 receptor that is located in Müller cells in rats.20 Further studies will investigate the molecular mechanisms of AGE induced neural apoptosis in the retina.
Abbreviations
AGE, advanced glycation end products
BSA, bovine serum albumin
DAPI, diamidino-phenyl-indole
DR, diabetic retinopathy
GCL, ganglion cell layer
GFAP, glial fibrillary acidic protein
GLAST, l-glutamate/l-aspartate transporter antibody
INL, inner nuclear layer
ONL, outer nuclear layer
OPL, outer plexiform layer
RAGE, AGE receptor
RPE, retinal pigment epithelium
TUNEL, terminal dUTP nick end labelling
VEGF, vascular endothelium growth factor
REFERENCES
- 1.Bloodworth JM Jr. Diabetic microangiopathy. Diabetes 1963;12:99–114. [DOI] [PubMed] [Google Scholar]
- 2.Shirao Y, Kawasaki K. Electrical responses from diabetic retina. Prog Retin Eye Res 1998;17:59–76. [DOI] [PubMed] [Google Scholar]
- 3.Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Prog Neuropsychopharmacol Biol Psychiatry 2003;27:283–90. [DOI] [PubMed] [Google Scholar]
- 4.Rungger-Brändle E, Dosso AA, Leuenberger PM. Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci 2000;41:1971–80. [PubMed] [Google Scholar]
- 5.Stitt A, Gardiner TA, Alderson NL, et al. The AGE inhibitor pyridoxamine inhibits development of retinopathy in experimental diabetes. Diabetes 2002;51:2826–32. [DOI] [PubMed] [Google Scholar]
- 6.Stitt AW. The role of advanced glycation in the pathogenesis of diabetic retinopathy. Exp Mol Pathol 2003;75:95–108. [DOI] [PubMed] [Google Scholar]
- 7.Takeuchi M, Bucala R, Suzuki T, et al. Neurotoxicity of advanced glycation end-products for cultured cortical neurons. J Neuropathol Exp Neurol 2000;59:1094–105. [DOI] [PubMed] [Google Scholar]
- 8.Hammes HP, Alt A, Niwa T, et al. Differential accumulation of advanced glycation end products in the course of diabetic retinopathy. Diabetologia 1999;42:728–36. [DOI] [PubMed] [Google Scholar]
- 9.Pinzon-Duarte G, Kohler K, Arango-Gonzales B, et al. Cell differentiation, synaptogenesis, and influence of the retinal pigment epithelium in a rat neonatal organotypic retinal culture. Vision Res 2000;40:3455–65. [DOI] [PubMed] [Google Scholar]
- 10.Vallazza-Deschamps G, Cia D, Gong J, et al. Excessive activation of cyclic nucleotide-gated channels contributes to neuronal degeneration of photoreceptors. Eur J Neurosci 2005; (in press). [DOI] [PubMed]
- 11.Lu M, Kuroki M, Amano S, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest 1998;101:1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono Y, Aoki S, Ohnishi K, et al. Increased serum levels of advanced glycation end-products and diabetic complications. Diabetes Res Clin Pract 1998;41:131–7. [DOI] [PubMed] [Google Scholar]
- 13.Picaud S, Peichl L, Franceschini N. Dye-induced photolesion in the mammalian retina: glial and neuronal reactions. J Neurosci Res 1993;35:629–42. [DOI] [PubMed] [Google Scholar]
- 14.Park SH, Park JW, Park SJ, et al. Apoptotic death of photoreceptors in the streptozotocin-induced diabetic rat retina. Diabetologia 2003;46:1260–8. [DOI] [PubMed] [Google Scholar]
- 15.Reber F, Geffarth R, Kasper M, et al. Graded sensitiveness of the various retinal neuron populations on the glyoxal-mediated formation of advanced glycation end products and ways of protection. Graefes Arch Clin Exp Ophthalmol 2003;241:213–25. [DOI] [PubMed] [Google Scholar]
- 16.Donovan M, Carmody RJ, Cotter TG. Light-induced photoreceptor apoptosis in vivo requires neuronal nitric-oxide synthase and guanylate cyclase activity and is caspase-3-independent. J Biol Chem 2001;276:23000–8. [DOI] [PubMed] [Google Scholar]
- 17.Duncan JL, LaVail MM, Yasumura D, et al. An RCS-like retinal dystrophy phenotype in Mer knockout mice. Invest Ophthalmol Vis Sci 2003;44:826–38. [DOI] [PubMed] [Google Scholar]
- 18.Liu BF, Miyata S, Kojima H, et al. Low phagocytic activity of resident peritoneal macrophages in diabetic mice: relevance to the formation of advanced glycation end products. Diabetes 1999;48:2074–82. [DOI] [PubMed] [Google Scholar]
- 19.Soulis T, Thallas V, Youssef S, et al. Advanced glycation end-products and their receptors co-localise in rat organs susceptible to diabetic microvascular injury. Diabetologia 1997;40:619–28. [DOI] [PubMed] [Google Scholar]
- 20.Uehara F, Ohba N, Ozawa M. Isolation and characterization of galectins in the mammalian retina. Invest Ophthalmol Vis Sci 2001;42:2164–72. [PubMed] [Google Scholar]