

SUMMARY
Ovol1 encodes a zinc finger transcriptional repressor that is downstream of the LEF1/β-catenin complex, nuclear effectors of canonical Wnt signaling. Previous gene knockout studies performed in a 129Sv x C57BL/6 mixed genetic background revealed that Ovol1-deficient mice survive to adulthood but display multiple tissue defects. In this study, we describe a C57BL/6 stain-specific reduction in perinatal survival of Ovol1 mutant mice. The perinatal lethality is accompanied by kidney epithelial cysts of embryonic onset and delayed skin barrier acquisition. Genetic analysis suggests a partial functional compensation by Ovol2 for the loss of Ovol1. The expression of Ovol2 was up-regulated in Ovol1-deficient epidermis, and Ovol1 represses the activity of Ovol2 promoter in a DNA binding-dependent manner. Collectively, these studies uncover novel functions of Ovol1 in mouse development and identify Ovol2 as a downstream target of Ovol1.
Keywords: Ovol1, Ovol2, perinatal lethality, skin barrier, cystic kidney, transcriptional repression
INTRODUCTION
Congenital abnormalities account for 11% of the perinatal death of human infants, a serious health issue especially in developing countries [1]. Understanding the genetic basis and tissue involvement of such abnormalities will ultimately facilitate the development of new and better treatment and management strategies to reduce mortality.
The evolutionarily conserved ovo genes encode transcription factors that either activate or repress gene expression and include members that reside downstream of developmental signaling pathways such as Wg/Wnt and BMP/TGF-β [2–4]. A common theme that emerges from functional studies of this gene family in various organisms is a requirement for the development and differentiation of epithelial tissues and germ cells [5–10]. While a single ovo gene exists in Drosophila and C. elegans, three ovo paralogs are found in mice, including Ovol1, Ovol2, and Ovol3 (also known as movo1, movo2, and movo3, respectively [3]). Ovol1 is expressed in multiple somatic epithelial tissues including skin and kidney, as well as in the male germinal epithelium [5]. Gene knockout studies revealed a functional requirement for Ovol1 in these expressing tissues, as mutant mice deficient in Ovol1 showed ruffled hairs, hyperproliferative epidermis, cystic kidneys, and defective spermatogenesis [5, 9, 11]. Despite such pleotropic defects, Ovol1 knockout mice maintained in a 129Sv x C57BL/6 (129 x B6) mixed genetic background [5, 9] survived to adulthood with no apparent compromise in life-span. Ovol2 however, is essential for embryonic development, as Ovol2-deficient mice die during mid-gestation [10]. Like Ovol1, Ovol2 is also expressed in adult epithelial tissues such as skin and kidney, and in the germinal epithelium of the testis [12].
It is not uncommon that genetic makeup affects the phenotypic manifestation of a particular mutation, and in fact, knockouts of several genes whose mutations affect epithelial development and physiology, including EGFR, keratin 8, and keratin 17, display phenotypic variations in different strain backgrounds [13–18]. Since the initial characterizations of Ovol1 mutant phenotypes were performed on a 129 x B6 mixed genetic background [5, 9], we transferred the Ovol1 mutant allele into a B6-enriched strain background in order to explore additional functions of Ovol1 in mammalian development. Our studies now reveal a novel function for Ovol1 in late embryonic and perinatal survival. Specifically, we show that Ovol1 mutant animals in the B6-enriched strain background die during late embryonic stages and within the first 2 weeks after birth. This compromised survival is accompanied by kidney cyst formation and delayed skin barrier development in the embryos. We present genetic data suggesting a functional compensation by Ovol2 for the loss of Ovol1 in perinatal survival. Finally, we provide molecular evidence demonstrating a direct negative regulation of Ovol2 expression by Ovol1.
MATERIALS AND METHODS
Mouse breeding and genotyping
Ovol1+/− mice in a 129 x B6 mixed genetic background were backcrossed with B6 mice for 8–10 sequential generations to create “congenic” mice containing the mutant allele (B6-Ovol1). B6-Ovol1+/− mice were then intercrossed, and offspring (embryos or pups) were genotyped as previously reported [5]. Ovol1+/−/Ovol2+/− double mutant mice were produced by crossing Ovol1+/− and Ovol2+/− single mutants in a B6-enriched background. The double heterozygotes were then intercrossed to generate compound mutants for analysis.
Histology, barrier assays, and immunohistochemistry
Embryos were fixed in Bouin’s fixative for 12–24 hours at room temperature, processed, and embedded in paraffin. Paraffin sections (5 μm) were prepared and stained with hematoxylin and eosin, and analyzed for morphology. Dye penetration assays were performed using freshly dissected embryos as described [19]. Tails and/or paws were removed for genotyping prior to staining. For immunohistochemistry, embryos were frozen in OCT, and sections (10 μm) were prepared and fixed with 4% paraformaldehyde for 10 minutes, followed by brief washes in PBS. Endogenous peroxidase was quenched by incubation in freshly prepared 3% H2O2 solution for 15 minutes. After three 5-minute washes in PBS, sections were subject to immunohistochemical analysis using a polyclonal antibody (1:50) against the full-length Ovol2 protein [20]. Images were acquired using a Nikon Eclipse E600 microscope.
Northern blot analysis
Total RNA was extracted from skin of 2.5-month old wild-type and B6-Ovol1 mutant mice, and northern blot analysis was performed as described [5] using a 320 bp PCR fragment containing sequences in exons 2 and 3 of Ovol2 as a cDNA probe.
Electrophoretic mobility shift assay (EMSA)
EMSA assays were performed using different amounts (as indicated in Figure legends) of partially purified recombinant His6-Ovol1 and ~ 20 fmols (~3 x 104 cpm) of gel-purified, 5’ 32P-end-labeled double-stranded oligonucleotides. Typically, binding reactions were carried out in 20 μl volume containing 20 mM Hepes (pH 7.9), 75 mM KCl, 2.5 mM MgCl2, 2 mM DTT, 1 mM EDTA, 12% glycerol and 1 μg of poly (dI-dC) for 30 minutes at room temperature. In competition experiments, a 20- or 100- fold molar excess of unlabeled specific or non-specific competitor was used. The protein-DNA complexes were resolved on 6% non-denaturing polyacrylamide gels and visualized by autoradiography.
Reporter assays
Assays were performed in 293T cells (a human kidney epithelial cell line) as previously described [9]. A typical transfection mixture contained a total of 0.5 μg of plasmids including 50 ng of pGL3-Ovol2 promoter construct or 10 ng of its mutant derivative with varying amounts of vectors (as indicated in figure legends) expressing Ovol1 [5] or VP16-Ovol1 [11], and 40 ng of a β-actin-β-gal construct. pCB-6 (+) (empty vector containing the CMV promoter) was used as stuffer DNA.
RESULTS
B6-Ovol1−/− mutants display perinatal lethality, cystic kidneys with a developmental onset, and delayed skin barrier acquisition
To explore novel functions of Ovol1 in mouse development, we transferred the previously generated Ovol1 mutant allele into a C57BL/6 (B6) strain background by 8–10 sequential backcrosses. While in the 129 x B6 mixed background homozygous mutants were born at the expected Mendelian frequency and survived to adulthood [5], intercrosses of Ovol1+/− mice enriched with the B6 genome (B6-Ovol1+/−) yielded fewer than expected Ovol1−/− pups when genotyped at postnatal day 14 (P14) (using χ2 test, p value is <0.005, indicating a <0.5% probability of conforming to the Mendelian law, which predicts a 1:2:1 ratio between +/+:+/−: −/−) (Table 1), implying embryonic or perinatal lethality of homozygous mutants in this new genetic background. To determine the timing of lethality, offspring from such intercrosses were collected at earlier stages and genotyped. Statistically significant deviations from the Mendelian law were observed for newborns and for embryos between the age of E15.5-E18.5, while approximate Mendelian ratios were obtained at and before E14.5 (Table 2). The step-wise decrease in the recovery of homozygous mutants from E15.5 to P14 suggests that death occurred both during late embryogenesis and in the first 2 postnatal weeks. The lethality was completely rescued by a single outcross of the B6-Ovol1 mice with CD1 mice (Table 1), confirming that the requirement for Ovol1 in embryonic/perinatal survival is specific to the B6 strain background.
Table 1.
Genotype distribution of P14 pups from Ovol1+/−intercrosses in different strain backgrounds
| Strain background | Genotype distribution | χ2 | p-value | ||
|---|---|---|---|---|---|
|
Ovol1+/+ |
Ovol1+/− |
Ovol1−/− |
|||
| 129xB6 | 27 | 51 | 25 | 0.1 | >0.9 |
| B6 congenic | 145 | 223 | 91 | 13.1 | <0.005 |
| CD1xB6 | 18 | 41 | 17 | 0.7 | >0.5 |
Table 2.
Genotype distribution of embryos and newborns from B6-Ovol1+/− intercrosses
| Age of offspring | Genotype distribution | χ2 | p-value | ||
|---|---|---|---|---|---|
|
Ovol1+/+ |
Ovol1+/− |
Ovol1−/− |
|||
| <E14.5 | 13 | 26 | 14 | 0.06 | >0.9 |
| E15.5–E18.5 | 144 | 197 | 120 | 12.2 | <0.005 |
| P0 | 117 | 146 | 87 | 14.8 | <0.005 |
The emergence of a novel lethality phenotype in a B6-enriched background implicates the presence of genetic modifier(s) of Ovol1 that functions differentially between B6 and 129 or CD1 strains. To address whether Ovol2 is such a modifier, we genotyped the surviving and perinatal lethal newborns for genetic markers flanking the Ovol2 locus (D2Mit285 and D2Mit395), but found no linkage between the Ovol2 locus and survival (data not shown). Therefore, it is unlikely that a B6 allele of Ovol2 is responsible for the phenotype aggravation of Ovol1 deletion or a 129 allele of Ovol2 is providing a protective allele.
Late embryonic and perinatal death can be caused by multiple defects such as developmental abnormalities in kidney (for example [21, 22]). Given our previous observation of a mild adult cystic kidney phenotype in the 129 x B6 mixed background [5], we wondered whether B6-Ovol1 mutant mice developed kidney abnormalities that are more severe or with an earlier age onset. Indeed, many B6-Ovol1−/− mutants showed severe unilateral or bilateral cystic kidneys that are apparent to the eye as early as 3 weeks postnatally (Fig. 1A, right), whereas in the original 129 X B6 background such severe cystic kidneys were detected in much older mutant animals and were always unilateral [23]. The penetrance of the defect is also much higher than the previous background, as most surviving B6-Ovol1−/− adults (>90%) displayed enlarged, fluid-filled kidneys. At a histological level, epithelial cysts were apparent as early as E17 in B6-Ovol1−/− embryos (Fig. 1B), whereas previously histological kidney anomalies were only detected postnatally [11]. No developmental defects were seen in kidneys from the B6-Ovol1+/− mice, and only one out of approximately 40 adult heterozygotes showed a unilateral cystic kidney.
Figure 1. Kidney cysts and delayed barrier acquisition in B6-Ovol1 mutant mice.
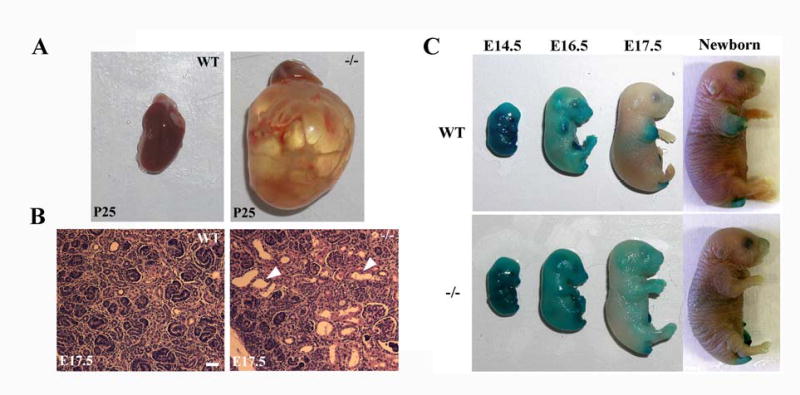
(A) Wild-type (left) and mutant (right) kidneys at postnatal day 25. (B) Histology of wild-type (left) and mutant (right) kidneys at E17.5. White arrowheads indicate epithelial cysts in the mutant kidney, while no such cysts were present in the wild-type control littermate. (C) Dye penetration assays of wild-type (top) and mutant (bottom) embryo at the indicated ages. Bar: 20μm in B.
Epidermal development during late embryogenesis gives rise to a functional skin permeability barrier composed of cross-linked proteins and lipids [24]. This barrier is essential for ex-utero survival of both mice and humans, as mice born with a defective barrier often die perinatally, whereas the onset of barrier formation in human embryos correlates with the lower age limit at which premature infants survive [24]. The lethality of B6-Ovol1 mutant mice and the known function of Ovol1 in developing epidermis [11] prompted us to use dye penetration assays to examine whether B6-Ovol1 mutants were defective in barrier development [19]. As expected, all wild-type embryos showed precocious sites of dye extrusion at E16.5, and were fully resistant to dye penetration by E17.5 (Fig. 1C, top). In contrast, more than half of the B6-Ovol1−/− embryos (n=8) showed no sign of barrier acquisition at E16.5, and all B6-Ovol1−/− E17.5 embryos examined (n=5) showed only partial dye extrusion (Fig. 1C, bottom). No apparent dye penetration defects were observed for B6-Ovol1−/− embryos at E18.5 and newborn (n=7), or for B6-Ovol1+/− embryos at all ages examined (data not shown). These results indicate that B6-Ovol1 homozygous mutant embryos were delayed in their barrier acquisition by approximately 1 day. Collectively, our findings reveal a previously unrecognized role for Ovol1 in embryonic/perinatal survival, and in embryonic kidney and skin barrier development.
Ovo12 expression is up-regulated in Ovol1-deficient epidermis and Ovol2 gene dosage affects the frequency of perinatal lethality of Ovol1-deficient mice
Functional compensation has been frequently observed between mammalian paralogs, and in such cases the expression of one paralog may be up-regulated when the other is disrupted. The incomplete penetrance of Ovol1−/−-associated perinatal lethality in the B6-enriched background led us to wonder whether Ovol2 might be up-regulated to compensate for the loss of Ovol1. Northern blot analysis indeed detected significantly higher levels of Ovol2 transcripts in skin from B6-Ovol1−/− mice than that from the wild-type littermates (Fig 2A). We also compared the expression of Ovol2 protein in wild-type and B6-Ovol1-deficient skin. In the wild-type epidermis, nuclear Ovol2 protein was detected in the proliferating basal layer, but not in differentiating suprabasal layers that are known to expression Ovol1 [5] (Fig. 2B). In Ovol1−/− epidermis however, nuclear Ovol2 protein was produced by not only basal but also suprabasal cells. Together, these results demonstrate that the disruption of Ovol1 in skin epidermis results in up-regulated expression of Ovol2, particularly in the suprabasal layers.
Figure 2. Up-regulated Ovol2 expression in Ovol1-deficient epidermis.
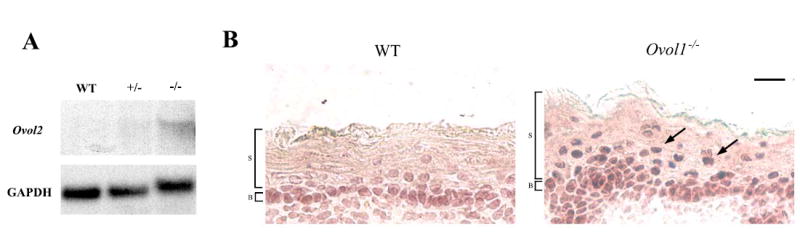
(A) Results of northern blot analysis on skin from a surviving 2.5 months-old Ovol1 −/− mouse and a control littermate. The same blot was stripped and reprobed with GAPDH as a loading control. (B) Results of immunohistochemistry of wild-type (left) and mutant (right) embryonic skin using a rabbit anti-Ovol2 antibody. Arrows indicate the presence of Ovol2 protein in suprabasal cells of the mutant. B, basal; S, suprabasal. Bar: 10 μm in B.
We previously generated Ovol2-deficient mice and observed embryonic death before E10.5 [10]. To examine whether Ovol2 provides a compensatory function for the loss of Ovol1, we first generated Ovol1+/−Ovol2+/− compound heterozygotes, which were fertile and displayed no overt phenotype (data not shown). We then intercrossed these mice and genotyped their pups to determine whether a reduction in the gene dosage of Ovol2 aggravated the postnatal lethality of Ovol1−/− mice. Consistent with the reported embryonic lethality caused by Ovol2 mutation [10], not a single double homozygous mutant was found at P14 among a total of 104 pups that were analyzed (data not shown). Regardless of the Ovol2 genotype, a less than expected number of Ovol1−/− pups were recovered, confirming our conclusion above that Ovol1 is required for optimum postnatal survival (Table 3). More importantly, a significantly lower-than-expected frequency of Ovol1−/−Ovol2+/− compound mutants was observed, as at a Mendelian ratio, Ovol1−/−Ovol2+/− : Ovol1−/−Ovol2+/+ should be 2:1 (Table 3; χ2= 5.08, p value is <0.025, indicating a <2.5% probability of conforming to this ratio). To examine whether the loss of both Ovol genes may lead to an earlier embryonic lethality than that are caused by the loss of Ovol2 alone, we also analyzed embryos from such intercrosses at E8.5. An expected frequency of occurrence was observed for Ovol1−/−Ovol2−/− embryos (5.75 was expected out of 92 embryos analyzed; 5 was observed), and these double mutants are morphologically identical to Ovol2−/− single mutants (data not shown). Collectively, these results indicate that the Ovol2 gene dosage affects the manifestation of the perinatal lethal phenotype of Ovol1-deficient animals, but argue against the notion that Ovol1 plays a redundant function for Ovol2 in early embryonic survival.
Table 3.
Genotype distribution of P14 pups from Ovol1+/−Ovol2+/− intercrosses
 |
 |
 |
||||
|---|---|---|---|---|---|---|
| Estimated | 8.7 | 17.4 | 17.4 | 34.8 | 8.7 | 17.4 |
| Observed | 12 | 20 | 21 | 34 | 9 | 8 |
| χ2 | 1.64 | 0.76 | 5.08 | |||
| p-value | >0.1 | >0.1 | <0.025 | |||
Molecular evidence that Ovol2 is a downstream target of Ovol1 transcriptional repression
We have previously reported that Ovol1 is a transcriptional repressor [9, 11]. The up-regulation of Ovol2 expression in Ovol1−/− suprabasal epidermal cells prompted us to test whether Ovol2 is a target of Ovol1 repression. We examined the sequence of an Ovol2 promoter fragment (1816 base pairs) that is able to direct active transcription in 293T cells (see below) for putative Ovol1 binding sites. Two CCGTTA elements, known to be the recognition consensus for Ovol1 [11], were found. Oligonucleotides Ovol2u and Ovol2d containing the upstream and downstream elements, respectively, were tested for Ovol1 binding using EMSA assays. Ovol1 bound to both Ovol2u and Ovol2d, and the sequence-specificity of the interaction was confirmed by competition by an excess amount of unlabeled specific (lanes 7, 8) but not non-specific (lanes 9, 10) oligonucleotides (Fig. 3A). Furthermore, the identity of the Ovol1-DNA complex was confirmed by the observation of a band supershift when anti-Ovol1 antibody was added to the binding reaction (Fig. 3A, lane 11). Therefore, Ovol2 promoter contains bona fide Ovol1 binding sites.
Figure 3. Ovol1 binds to sequences in the Ovol2 promoter and represses its activity in reporter assays.
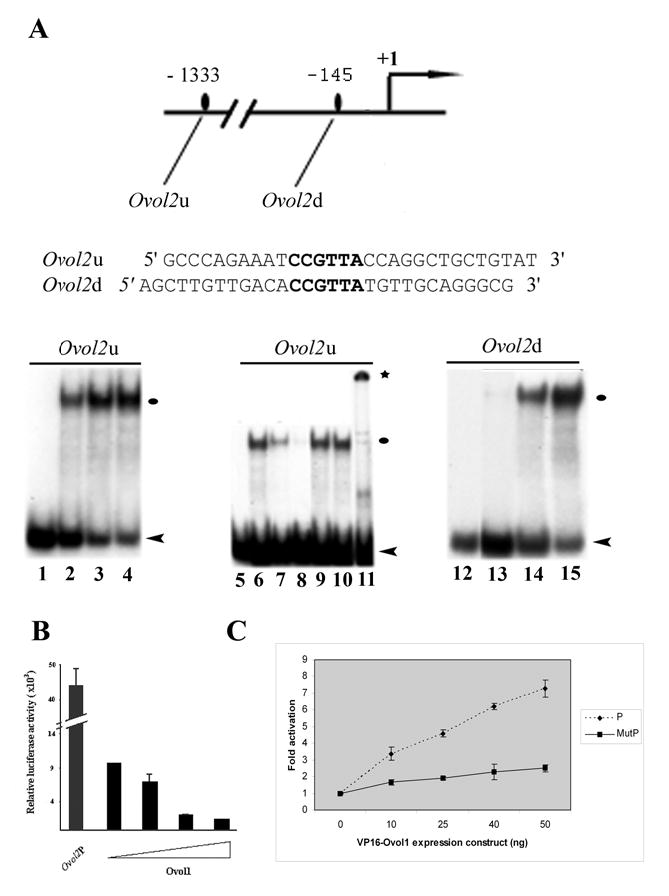
(A) EMSA assays were carried out with the Ovol2u (left) and Ovol2d (right) oligonucleotides. Lanes 1, 5, 12: no protein added. Lanes 2, 6, 13: with 0.2 μg recombinant Ovol1. Lanes 3 and 14: with 0.3 μg recombinant Ovol1. Lanes 4 and 15: with 0.5 μg recombinant Ovol1. Lanes 7–11: same as lane 6 but in the presence of a 20- (lanes 7, 9) or 100-fold (lanes 8, 10) excess of unlabeled specific (lanes 7, 8) or non-specific (lanes 9, 10) oligonucleotides, or in the presence of an anti-Ovol1 antibody (lane 11). Black ovals and * indicate the Ovol1-DNA and Ovol1-DNA-antibody complexes, respectively. Arrowheads indicate the position of the free probes. (B–C) Repression (B) and activation (C) of Ovol2 promoter by Ovol1 (B) and VP16-Ovol1 (C), respectively. The triangle in B indicates increasing concentrations (0, 0.01 μg, 0.02 μg, and 0.05 μg) of the Ovol1expression vector. P, wild-type promoter; MutP, mutant promoter in which the Ovol1 binding sites are disrupted. Each bar represents the average of triplicate samples in a single experiment, and results are representative of several independent experiments. Luciferase activities were normalized for transfection efficiency by using a β-actin promoter driving lacZ as an internal control.
We next used reporter assays to examine the effect of Ovol1 on Ovol2 promoter activity. To do this, we cloned the Ovol2 promoter fragment upstream of a luciferase reporter gene. Co-transfection with an Ovol1 expression vector led to a dosage-dependent repression of the activity of this promoter (Fig. 3B). Since repression can sometimes result as an artifact from high levels of protein expression, we turned to test whether VP16-Ovol1, a fusion protein that was shown to activate known Ovol1 target genes [9, 11], can activate the Ovol2 promoter. This fusion protein indeed activated the Ovol2 promoter in a dosage-dependent manner (Fig. 3C), while VP16 alone had no effect (data not shown). To address whether the observed regulation of Ovol2 promoter by Ovol1 depends on Ovol1 binding to the promoter, we repeated the experiments, this time using a mutant promoter where both Ovol1 binding sites were removed (by replacing the CCGTTA sequence with a hexamer to which Ovol1 does not bind; [11]). A significant reduction in the degree of activation by VP16-Ovol1 was observed (Fig. 3D), indicating that the maximum effect of Ovol1 on Ovol2 promoter activity requires Ovol1 binding to these sites. Collectively, these results demonstrate that Ovol1 represses Ovol2 transcription by binding to its promoter.
DISCUSSION
Our studies have identified a novel function of Ovol1 in late embryonic/postnatal survival. Furthermore, these results demonstrate that the manifestation of this function is dependent on the strain background where the Ovol1 mutant alleles reside. There has been an increasing recognition that many Mendelian traits that result in birth defects or adult diseases in mice or human vary in their phenotypic manifestations due to the presence of genetic modifiers [25]. Our work now adds Ovol1 to the list of genes, including EGFR, keratin 8, and keratin 17, the function of which is influenced by strain-specific modifier(s) [13–18]. Future work to identify such modifier(s) offers exciting opportunities to unravel novel genetic players or pathways that govern developmental processes in which Ovol1 is required.
What is the underlying basis of perinatal lethality of Ovol1 knockout mice? While we do not have a definitive answer to this question, we observed defects that are known to associate with perinatal lethality. All mutant embryos examined were able to extrude dye by birth despite a developmental delay in barrier acquisition, making it unlikely that a barrier defect is the underlying cause of lethality. This said, we note that the frequency of a delayed barrier (approximately half) in Ovol1−/− embryos is intriguingly similar to that of postnatal lethality of Ovol1−/− pups. Furthermore, half of the Ovol1−/− pups contain cornified envelops, important constituents of the actual barrier, that show lower resistance to mechanical stress (data not shown). Therefore, we cannot fully exclude the possibility that a compromised barrier under environmental stresses contributes to a certain extent to the reduced ex utero survival of Ovol1 mutant animals. Kidney defect is likely an important contributing factor to lethality. The developmental onset of kidney cysts in B6-Ovol1−/− animals is reminiscent of that observed in mice mutant for PKD1, a major target of cystic kidney diseases in humans [22]. PKD1-deficient mice die perinatally, presenting an interesting parallel with the B6-Ovol1−/− mice. Most known proteins whose mutations cause cystic renal diseases are localized to the primary cilium of renal tubular epithelial cells [26, 27]. Our finding that deletion of Ovol1, a regulatory instead of structural protein, also causes kidney cysts is a first. Our B6-Ovol1−/− mice should now provide a useful animal model to study the regulatory mechanisms underlying kidney development and the molecular basis of human cystic kidney diseases.
We used genetic approaches to address the possibility that Ovol2 provides a compensatory function for the loss of Ovol1. We found that mice with only the two Ovol1 alleles disrupted (Ovol1−/−Ovol2+/+) survived better than those with both Ovol1 alleles as well as a copy of the Ovol2 allele disrupted (Ovol1−/−Ovol2+/−). Taking this result together with our observation of increased Ovol2 expression in an Ovol1-deficient background, we surmise that the elevated expression of Ovol2 compensates for the loss of Ovol1, and that this compensation is insufficient with only one wild-type copy of Ovol2. Based on this finding, we predict that a complete penetrance of perinatal lethality and/or additional phenotypes might surface when both alleles of Ovol2 are deleted in the Ovol1-deficient background. Since Ovol2 homozygous deletion causes embryonic lethality prior to the stage when Ovol1 function is required, a conditional deletion of Ovol2 needs to be performed to address this notion.
The existing model suggests that Ovol1 functions to regulate the transition of epithelial progenitor cells from proliferation to terminal differentiation possibly by regulating the expression of downstream target genes including c-Myc and Id2 [9, 11]. Our in vivo and in vitro studies now add Ovol2 to the list of Ovol1 downstream targets. Under physiological conditions, Ovol2 is expressed in the proliferating basal but not differentiating suprabasal cells of the epidermis. This expression pattern is reminiscent of c-Myc and Id2, known positive regulators of proliferation and negative regulators of differentiation. Does Ovol2 function similarly like c-Myc or Id2 to positively regulate proliferation and negatively regulate terminal differentiation? While this may be the case in normal epidermis, the observed compensation by Ovol2 for the loss of Ovol1 suggests that the situation under diseased conditions (e.g., when Ovol1 is lost) is more complex. Clearly, additional studies are warranted to understand the role of Ovol2 in epidermal proliferation and differentiation under physiological and pathological conditions.
Acknowledgments
We thank Magid Fallahi and Ming Hu for help with mouse breeding. This work was supported by NIH Grants R01-AR47320 and K02-AR51482 awarded to X. D., and Mahalakshmi Nair was partially supported by an institutional predoctoral NIH Training Program in Developmental Mechanisms Underlying Congenital Defects.
References
- 1.Sule SS, Onayade AA. Community-based antenatal and perinatal interventions and newborn survival. Niger J Med. 2006;15:108–14. doi: 10.4314/njm.v15i2.37091. [DOI] [PubMed] [Google Scholar]
- 2.Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A. Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol. 2004;24:4241–54. doi: 10.1128/MCB.24.10.4241-4254.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li B, Mackay DR, Dai Q, Li TWH, Nair M, Fallahi M, Schonbaum C, Fantes J, Mahowald A, Waterman ML, Fuchs E, Dai X. The LEF1/β-catenin complex activates movo1, a mouse homolog of Drosophila ovo gene required for epidermal appendage differentiation. Proc Natl Acad Sci USA. 2002;99:6064–6069. doi: 10.1073/pnas.092137099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Payre F, Vincent A, Carreno S. ovo/svb integrates Wingless and DER pathways to control epidermis differentiation. Nature. 1999;400:271–5. doi: 10.1038/22330. [DOI] [PubMed] [Google Scholar]
- 5.Dai X, Schonbaum C, Degenstein L, Bai W, Mahowald A, Fuchs E. The ovo gene required for cuticle formation and oogenesis in flies is involved in hair formation and spermatogenesis in mice. Genes Dev. 1998;12:3452–63. doi: 10.1101/gad.12.21.3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson AD, Fitzsimmons D, Hagman J, Chamberlin HM. EGL-38 Pax regulates the ovo-related gene lin-48 during Caenorhabditis elegans organ development. Development. 2001;128:2857–65. doi: 10.1242/dev.128.15.2857. [DOI] [PubMed] [Google Scholar]
- 7.Mevel-Ninio M, Terracol R, Salles C, Vincent A, Payre F. ovo, a Drosophila gene required for ovarian development, is specifically expressed in the germline and shares most of its coding sequences with shavenbaby, a gene involved in embryo patterning. Mech Dev. 1995;49:83–95. doi: 10.1016/0925-4773(94)00305-7. [DOI] [PubMed] [Google Scholar]
- 8.Oliver B, Perrimon N, Mahowald AP. The ovo locus is required for sex-specific germ line maintenance in Drosophila. Genes Dev. 1987;1:913–23. doi: 10.1101/gad.1.9.913. [DOI] [PubMed] [Google Scholar]
- 9.Li B, Nair M, Mackay DR, Bilanchone V, Hu M, Fallahi M, Song H, Dai Q, Cohen PE, Dai X. Ovol1 regulates meiotic pachytene progression during spermatogenesis by repressing Id2 expression. Development. 2005;132:1463–73. doi: 10.1242/dev.01658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mackay DR, Hu M, Li B, Rheaume C, Dai X. The mouse Ovol2 gene is required for cranial neural tube development. Dev Biol. 2006;291:38–52. doi: 10.1016/j.ydbio.2005.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nair M, Teng A, Bilanchone V, Agrawal A, Li B, Dai X. Ovol1 regulates the growth arrest of embryonic epidermal progenitor cells and represses c-myc transcription. J Cell Biol. 2006;173:253–64. doi: 10.1083/jcb.200508196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li B, Dai Q, Li L, Nair M, Mackay D, Dai X. Ovol2, a Mammalian Homolog of Drosophila ovo: Gene Structure, Chromosomal Mapping, and Aberrant Expression in Blind-Sterile Mice. Genomics. 2002;80:319. doi: 10.1006/geno.2002.6831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Threadgill DW, Dlugosz AA, Hansen LA, Tennenbaum T, Lichti U, Yee D, LaMantia C, Mourton T, Herrup K, Harris RC, et al. Targeted disruption of mouse EGF receptor: effect of genetic background on mutant phenotype. Science. 1995;269:230–4. doi: 10.1126/science.7618084. [DOI] [PubMed] [Google Scholar]
- 14.Sibilia M, Wagner EF. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science. 1995;269:234–8. doi: 10.1126/science.7618085. [DOI] [PubMed] [Google Scholar]
- 15.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Werb Z, Derynck R. Epithelial immaturity and multiorgan failure in mice lacking epidermal growth factor receptor. Nature. 1995;376:337–41. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]
- 16.Baribault H, Penner J, Iozzo RV, Wilson-Heiner M. Colorectal hyperplasia and inflammation in keratin 8-deficient FVB/N mice. Genes Dev. 1994;8:2964–73. doi: 10.1101/gad.8.24.2964. [DOI] [PubMed] [Google Scholar]
- 17.Baribault H, Price J, Miyai K, Oshima RG. Mid-gestational lethality in mice lacking keratin 8. Genes Dev. 1993;7:1191–202. doi: 10.1101/gad.7.7a.1191. [DOI] [PubMed] [Google Scholar]
- 18.McGowan KM, Tong X, Colucci-Guyon E, Langa F, Babinet C, Coulombe PA. Keratin 17 null mice exhibit age- and strain-dependent alopecia. Genes Dev. 2002;16:1412–22. doi: 10.1101/gad.979502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hardman MJ, Sisi P, Banbury DN, Byrne C. Patterned acquisition of skin barrier function during development. Development. 1998;125:1541–52. doi: 10.1242/dev.125.8.1541. [DOI] [PubMed] [Google Scholar]
- 20.Mackay DR. Ph.D. thesis. University of California; Irvine: 2005. [Google Scholar]
- 21.Moser M, Pscherer A, Roth C, Becker J, Mucher G, Zerres K, Dixkens C, Weis J, Guay-Woodford L, Buettner R, Fassler R. Enhanced apoptotic cell death of renal epithelial cells in mice lacking transcription factor AP-2beta. Genes Dev. 1997;11:1938–48. doi: 10.1101/gad.11.15.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu W, Peissel B, Babakhanlou H, Pavlova A, Geng L, Fan X, Larson C, Brent G, Zhou J. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat Genet. 1997;17:179–81. doi: 10.1038/ng1097-179. [DOI] [PubMed] [Google Scholar]
- 23.Dai X, Segre JA. Transcriptional control of epidermal specification and differentiation. Curr Opin Genet Dev. 2004;14:485–91. doi: 10.1016/j.gde.2004.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Segre J. Complex redundancy to build a simple epidermal permeability barrier. Curr Opin Cell Biol. 2003;15:776–82. doi: 10.1016/j.ceb.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Nadeau JH. Modifier genes in mice and humans. Nat Rev Genet. 2001;2:165–74. doi: 10.1038/35056009. [DOI] [PubMed] [Google Scholar]
- 26.Vogel G. News focus: Betting on cilia. Science. 2005;310:216–8. doi: 10.1126/science.310.5746.216. [DOI] [PubMed] [Google Scholar]
- 27.Witzgall R. New developments in the field of cystic kidney diseases. Curr Mol Med. 2005;5:455–65. doi: 10.2174/1566524054553496. [DOI] [PubMed] [Google Scholar]