

Abstract
Hepatocytes from cirrhotic murine livers exhibit increased basal ROS activity and resistance to TGFβ-induced apoptosis, yet when ROS levels are decreased by antioxidant pretreatment, these cells recover susceptibility to apoptotic stimuli. To further study these redox events, hepatocytes from cirrhotic murine livers were pretreated with various antioxidants prior to TGFβ treatment and the ROS activity, apoptotic response, and mitochondrial ROS generation were assessed. In addition, normal hepatocytes were treated with low-dose H2O2 and ROS and apoptotic responses determined. Treatment of cirrhotic hepatocytes with various antioxidants decreased basal ROS and rendered them susceptible to apoptosis. Examination of normal hepatocytes by confocal microscopy demonstrated co-localization of ROS activity and respiring mitochondria. Basal assessment of cirrhotic hepatocytes showed non-focal ROS activity that was abolished by antioxidants. After pretreatment with an adenovirus expressing MnSOD, basal cirrhotic hepatocyte ROS was decreased and TGFβ-induced co-localization of ROS and mitochondrial respiration was present. Treatment of normal hepatocytes with H2O2 resulted in a sustained increase in ROS and resistance to TGFβ apoptosis that was reversed when these cells were pretreated with an antioxidant. In conclusion, cirrhotic hepatocytes have a non-focal distribution of ROS. However, normal and cirrhotic hepatocytes exhibit mitochondrial localization of ROS that is necessary for apoptosis.
Keywords: Reactive oxygen species (ROS), hepatocytes, apoptosis, transforming growth factor beta (TGFβ), mitochondria
List of Abbreviations: AdCat: Adenovirus expressing catalase; AdLuc: Adenovirus expressing luciferase; AdMnSOD: Adenovirus expressing MnSOD; DMNQ: 2,3-dimethoxy-1,4-naphthoquinone; H2-DCFDA: 2’,7’-dichlorofluorescein diacetate; ROS: Reactive oxygen species; TGFβ: Transforming growth factor beta 1
INTRODUCTION
Transforming growth factor beta (TGFβ) induces apoptosis in normal murine hepatocytes through an apoptotic pathway that requires reactive oxygen species (ROS) generation, the mitochondrial permeability transition (MPT) with cytochrome c release, and caspase activation [1]. The increase in ROS after TGFβ is an early event that occurs within 90 minutes, lasts approximately three hours, and precedes the MPT and caspase activation [1,2]. Furthermore, inhibition of a ROS burst abolishes the apoptotic response and related intracellular events [1–3]. The source and mechanism of TGFβ-induced ROS has been attributed to the mitochondria, microsomes, and membrane-associated NADPH oxidase-like systems, yet the evanescent nature of ROS has made definitive source identification difficult [4,5]. In addition, TGFβ-induced down-regulation of the anti-oxidant, glutathione, further complicates the balance of ROS production versus scavenger activity [4,6]. Therefore, although ROS play an integral role in hepatocyte death following TGFβ administration, the necessity of ROS and the intracellular mechanisms through which ROS-mediated events occur remain unclear.
Despite the requirement of ROS generation for TGFβ-induced hepatocyte apoptosis in normal cells, increased intracellular ROS in chronic inflammatory states does not inevitably induce parenchymal cell death, and, in fact, may allow an adaptive state that protects against cell death [1]. Previous work demonstrated that in a carbon tetrachloride (CCl4)-induced murine model of liver cirrhosis, hepatocytes isolated from this chronically inflamed liver have a greater than 1.5-fold increase in ROS under basal conditions, fail to generate a ROS burst in response to TGFβ, resist apoptosis, yet upon pretreatment with the anti-oxidant, trolox, recovered responsiveness to TGFβ-induced programmed cell death [1]. The association between increased cellular ROS and resistance to cell death has been noted not only in chronic inflammatory conditions, but also in neoplastic cells and changes in ROS may be associated with a malignant phenotype [7,8]. The source of ROS generation in chronic inflammation and neoplasia is unknown in these disease states in which chronic hypoxia may instigate free radical generation. Moreover, initiation of a single oxygen-derived free radical pathway within a given cellular locale can propagate rapidly and exponentially to multiple intertwined oxidant generating pathways within various cellular compartments thereby rendering identification of the primary ROS generating pathway difficult.
The cellular ROS state represents the balance of free radical production and maintenance versus anti-oxidant scavenging activity, and, therefore, the cellular expression of anti-oxidant enzymes such as the catalase, superoxide dismutases (SOD) 1 and 2, and the glutathione peroxidase systems should be examined in chronic inflammation and neoplasia. Previous studies have documented decreased anti-oxidant gene expression both in non-inflammatory and in inflammatory and neoplastic conditions [4]. Furthermore, other studies have suggested that anti-oxidants such as SOD2 (MnSOD) may act as tumor suppressors by controlling the cellular ROS state [8]. Because the expression of anti-oxidant enzymes at the time of ROS generation is often unknown, it is difficult to discern if decreased anti-oxidant expression is the cause of or results from increased ROS [4]. Furthermore, the variability in quantifying ROS at a given time and the relative specificity of various exogenous anti-oxidants for oxidative pathways leads one to question the importance of decreased anti-oxidants as a primary cause for the increased ROS in disease state like inflammation and neoplasia.
Because our previous data [1] suggested that an increase in ROS resulted in TGFβ-induced apoptosis in normal hepatocytes but prevented apoptosis in hepatocytes from a cirrhotic liver, we sought to examine these hepatocytes from normal and cirrhotic livers to further elucidate the importance of ROS in hepatocyte responsiveness to pro-apoptotic stimuli such as TGFβ. We found that in normal hepatocytes, TGFβ-induced ROS was initiated in the mitochondria and that inhibition of ROS precluded apoptosis. In addition, under basal conditions, cirrhotic hepatocytes demonstrated a diffuse increase in ROS which was abolished with trolox and in response to TGFβ these anti-oxidant treated cells demonstrated an acute increase in mitochondrial derived ROS. Finally, normal hepatocytes treated with low-dose H2O2 developed a sustained increase in ROS which inhibited TGFβ-induced apoptosis in these converted hepatocytes.
MATERIALS AND METHODS
Materials
Adult, eight week-old, male BALB/c mice were obtained from Harlan Laboratories (Indianapolis, IN). Deferoxamine, 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), glutathione, N, N-Diphenyl-1,4-phenylenediamine (DPPD), N-acetylcysteine (NAC), and trolox were acquired from Sigma Chemical (St. Louis, MO). Catalase adenovirus (AdCat) was a kind gift from the University of Iowa Vector Core. SOD2 (AdMnSOD) and luciferase (AdLuc) adenoviruses were obtained from the UNC Chapel Hill Vector Core. Anti-caspase 3 rabbit polyclonal antibody was purchased from Cell Signaling Technology (Beverly, MA). 2’, 7’-dichlorofluorescein diacetate (H2-DCFDA) and MitoTracker Red (MTR) were obtained from Molecular Probes (Eugene, OR).
Hepatocyte Isolation and Culture
Eight week-old, male BALB/c mice weighing 20–25 grams were injected in the peritoneum twice-weekly with 2 ml/kg of 50% carbon tetrachloride (CCl4; cirrhotic mice) in sterile mineral oil or an equal volume of mineral oil alone for a total of eight weeks [1]. All hepatocyte isolations were performed following a 7-day recovery. Hepatocytes were isolated through an abdominal incision that allowed cannulation of the inferior vena cava, clamping of the suprahepatic inferior vena cava, and transection of the portal vein. The liver was perfused in retrograde fashion through the hepatic veins. Initially, the liver was perfused with a solution containing 0.25 M HEPES, 115 mM NaCl, 50mM KCl, 10mM KH2PO4, and 0.5 mM EGTA at pH 7.4 for a total volume of 50–100 ml. The perfusate was changed to a solution without EGTA but containing 1mM CaCl2 and 0.4 mg/ml type I collagenase (0.6 mg/ml for cirrhotic livers, Worthington Biochemical. Lakewood, NJ) at pH 7.4 for a total volume of 100–150 ml. The liver was excised, combed manually to disperse the cells, and subjected to differential centrifugation. Cell viability was determined using Trypan blue exclusion with a viability of >90% accepted for experiments. Hepatocytes were plated in Waymouth’s medium supplemented with 10% fetal calf serum, 5 μg/ml insulin, and 100 nmol/L dexamethasone. After two hours, the medium was changed to hormonally defined medium (HDM) containing insulin (5 μg/ml, Sigma Biochemical, St. Louis, MO), transferrin (5 μg/ml), selenium (30 nM), and free fatty acid (1.52 μM of palmitic acid, palmitoleic acid, stearic acid, oleic acid, linoleic acid and linolenic acid, Sigma Biochemical, St. Louis, MO).
Adenovirus Purification and Infection
The replication-deficient adenoviruses expressing either the luciferase (AdLuc), as a control, catalase (AdCat), or manganese superoxide dismutase (AdMnSOD) were prepared and stored as described previously [1]. Twenty-four hours prior to treatment control and cirrhotic hepatocytes were infected at a multiplicity of infection of 100. Expression of the transgene was confirmed by immunoblot or luciferase assay and transfection efficiency was routinely greater than 80%.
Morphologic Assessment of Apoptosis
Propidium iodide (PI) staining and fluorescent microscopy was used for morphologic assessment of apoptosis [1]. Hepatocytes were fixed with methanol-acetic acid (3:1) for 10 minutes at 4°C, washed twice, and stained with PI (0.33 mg/ml) and visualized under green excitation light using a IX-Olympus microscope (Olympus, Tokyo, Japan). The number of condensed nuclei in five high-powered fields (X400) was determined as a percent of the total number of nuclei.
ROS Determination
Cells were assayed in triplicate at a density of 1x105 per well in a 12-well plate and after removing media one ml H2-DCFDA solution (10 μM in DMSO) was added and incubated at 37°C for 20 minutes. H2O2-treated cells served as positive controls. Fluorescence was determined in a Fluostar Spectrofluorometer (BMG Labtech, Durham, NC) and read with wavelengths of excitation of 488 nm and emission of 525 nm, respectively. Cell lysates were the harvested for determination of protein concentration using the Bradford assay.
Immunoblot Analysis
To obtain whole cell extracts, cells were rinsed twice in PBS and lysed in buffer containing 0.05 M Tris, pH 7.3, 0.15 M NaCl, 1% NP40, 0.5% deoxycholate, and the protease inhibitor cocktail (Sigma) for 10 minutes at 4°C. Samples were centrifuged at 14,000 rpm to remove debris and the protein concentration was determined by Bradford assay. Following SDS-polyacrylamide gel electrophoresis, samples were transferred to PVDF membranes and blocked in 5% non-fat milk in TBS-T. After a one hour incubation with primary antibodies at concentrations recommended by the manufacturers, blots were washed for 15 minutes in TBS-T, incubated with HRP-conjugated secondary antibody (1:1000) for 30 minutes, washed for 15 minutes in TBS-T and visualized by enhanced chemiluminescence (Amersham Biosciences, Piscataway, NJ) and exposed to a Biomax-MS film (PerkinElmer, Boston, MA).
Caspase Activity
Caspase-3 activity was assessed by a colorimetric enzyme assay (BD Biosciences, San Jose, CA) [1]. Each assay was performed in triplicate with 2x106 hepatocytes. Cell lysates from treated or untreated hepatocytes were incubated with 50 μl of 2X reaction buffer/DTT and 5 μl of 1 mM caspase-3 substrate (DEVD-pNA) was added. Following incubation at 37°C for one hour absorbance was determined at 405 nm in a spectrophotometer.
Confocal Microscopy
Localization of TGFβ-induced ROS production to mitochondria was investigated in hepatocytes dual-labeled with H2-DCFDA (green fluorescence) to detect ROS and MitoTracker Red (MTR; Molecular Probes, Eugene, OR) to detect mitochondria [1]. MTR is electrophoretically taken up by the mitochondria. After uptake, MTR becomes covalently bound to sulfhydryl groups of mitochondrial proteins and remains in the mitochondria even if the mitochondrial depolarize [9,10]. Co-localization of these signals yields a yellow signal that indicates mitochondria actively producing ROS. Hepatocytes, 3 X 105, were plated overnight in a MatTek culture dish (MatTek, Ashland, MA) and treated or untreated for 20 minutes at 37°C and H2-DCFDA and MTR were added to achieve final concentrations of 2 μM and 500 nM, respectively. After one hour incubation, the medium were aspirated, hepatocytes were subjected to two wash steps with PBS, and confocal imaging performed. Confocal images were obtained with a Zeiss LSM 510 laser scanning confocal microscope (Thornwood, NY). Detector gain was equal in both experimental and control groups. Cells were randomly selected and analyzed for co-localization of H2-DCF fluorescence and mitochondrial staining with MTR. The images of green H2-DCF and red MTR were superimposed and distinct regions of yellow provided direct evidence of mitochondrial derived ROS. The detector gains used on the confocal microscope for both the dyes were selected so as to not obscure visualization of subcellular features of the cell while still allowing discrimination of signals proportional to activity. These gain levels were not altered during the course of the experiments. However, by keeping gains constant, MTR intensity levels varied slightly between the experiments. Images were background corrected before analysis and average fluorescence was calculated using Adobe Photoshop (San Jose, CA).
H2O2-Treated Hepatocytes
Normal hepatocytes were treated with 10 μM H2O2 for 10–20 minutes and ROS activity assessed with H2-DCF for 72 hours. ROS activity in these hepatocytes was increased approximately two-fold and maintained this level of activity for 72 hours. This ROS activity is similar to that seen in cirrhotic hepatocytes at baseline. These cells were then subjected to TGFβ treatment and apoptosis determined at 48 hours as described previously.
Determination of Protein Tyrosine Phosphatase Activity
Protein tyrosine phosphatase activity was spectrometrically determined using p-nitrophenyl phosphate [11].
Data Analysis
All experiments were performed in at least triplicate. Data are reported as mean plus or minus the standard deviation. Statistical analysis was performed using ANOVA and Dunnett’s t-test and a p-value of 0.05 was considered significant.
RESULTS
Exogenous Antioxidants and Cirrhotic Hepatocyte Apoptosis
Previous work from our laboratory demonstrated that cirrhotic hepatocyte resistance to TGFβ-induced apoptosis was mediated through ROS and that pretreatment with trolox decreased ROS and rendered these hepatocytes susceptible to TGFβ-induced apoptosis [1]. To determine if antioxidants selective for specific ROS generating pathways would provide information about the source of ROS production in cirrhotic hepatocytes, these cells were treated with 1.5 mM deferoxamine, 5 mM glutathione, 5 μM DPPD, or 2.5 mM NAC for 1 hour. Each treatment group exhibited a significant decrease that returned ROS to control levels in response to the various antioxidants (data not shown). Furthermore, following antioxidant pretreatment each group demonstrated a significant increase in ROS generation after TGFβ administration (Fig. 1A). This ROS spike 90 minutes after TGFβ administration corresponds to generation of a ROS burst that occurs in normal hepatocytes [1]. Importantly, the ROS burst following TGFβ administration in cirrhotic hepatocytes pretreated with the various antioxidants was associated with a significant increase in morphologic apoptosis at 48 hours (Fig. 1B). These data suggest that cirrhotic hepatocytes respond to several exogenous antioxidants with decreased ROS and that a reduction in baseline ROS in cirrhotic hepatocytes permits a TGFβ-induced ROS burst with subsequent apoptosis. However, exogenous administration of multiple antioxidants does not specify the ROS initiating pathway in cirrhotic hepatocytes.
Figure 1.
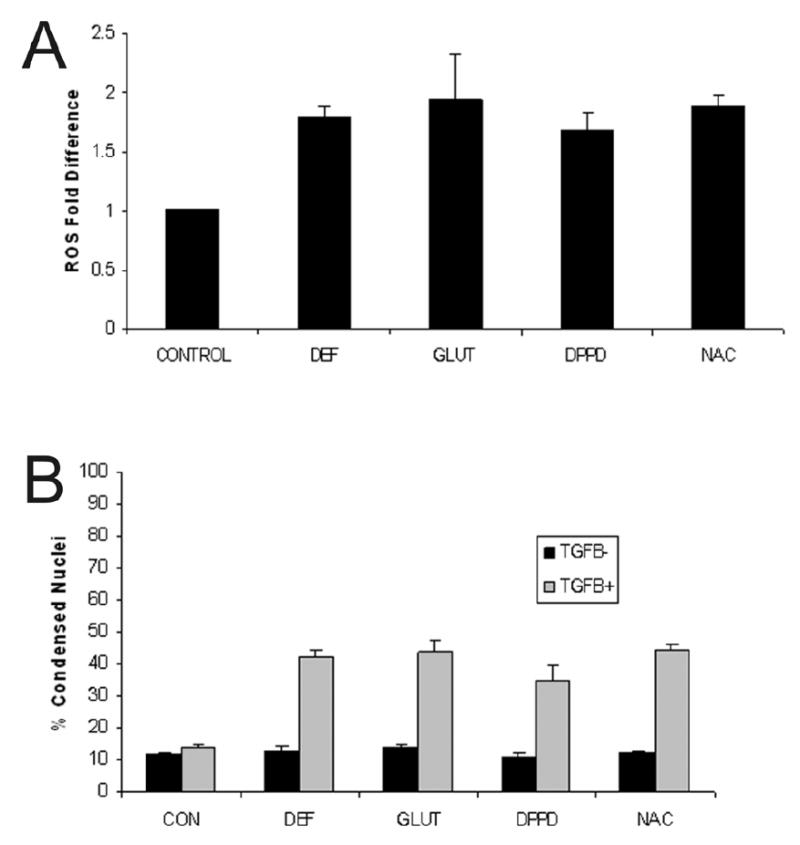
(A) Cirrhotic hepatocytes were pretreated with exogenous antioxidants including deferoxamine (DEF; 1.5mM), glutathione (GLUT; 5 mM), DPPD (5 μM), or N-acetylcysteine (NAC; 2.5 mM) for one hour prior to administration of TGFβ (5 ng/ml) and ROS activity measured 90 minutes after TGFβ administration. The ROS activity, expressed as fold-difference, returned to baseline at time zero (data not shown), but increased in response to TGFβ treatment for all antioxidants tested. These findings suggest involvement of multiple ROS generating pathways in increased cirrhotic hepatocyte basal ROS activity. (B) The percent of condensed nuclei, indicative of morphologic apoptosis, was determined after cirrhotic hepatocytes were pretreated with exogenous antioxidants (agents and dose identical to Fig. 1) for one hour prior to treatment with or without TGFβ (5 ng/ml) and apoptosis determined at 48 hours. Pretreatment with the antioxidants alone did not induced apoptosis, but antioxidant treatment followed by TGFβ administration increased markedly cirrhotic hepatocyte apoptosis for each antioxidant tested.
Adenovirus Expression of Antioxidants and Cirrhotic Hepatocyte Apoptosis
To determine if adenoviruses expressing antioxidant enzymes had an effect on cirrhotic hepatocytes similar to exogenously applied antioxidants, adenoviruses expressing luciferase as a control (AdLuc), catalase (AdCat), or MnSOD (AdMnSOD) were administered. The adenoviruses expressing MnSOD and catalase both decreased ROS in cirrhotic hepatocytes (Fig. 2A), and these cells were capable of attaining a ROS spike 90 minutes following TGFβ treatment (Fig. 2B). The adenovirus expressing MnSOD was particularly effective in decreasing ROS and permitting a robust ROS response to TGFβ. In addition, assessment of apoptosis (Fig. 2C) demonstrated that cirrhotic hepatocytes transduced with either the MnSOD or catalase adenoviruses underwent significant apoptosis 48 hours following treatment with TGFβ. These findings indicate that cirrhotic hepatocytes infected with antioxidant enzymes behave similarly to cirrhotic hepatocytes treated with exogenous antioxidants; however, adenoviral expression of MnSOD appeared particularly effective in reducing ROS and permitting a TGFβ-induced ROS burst.
Figure 2.
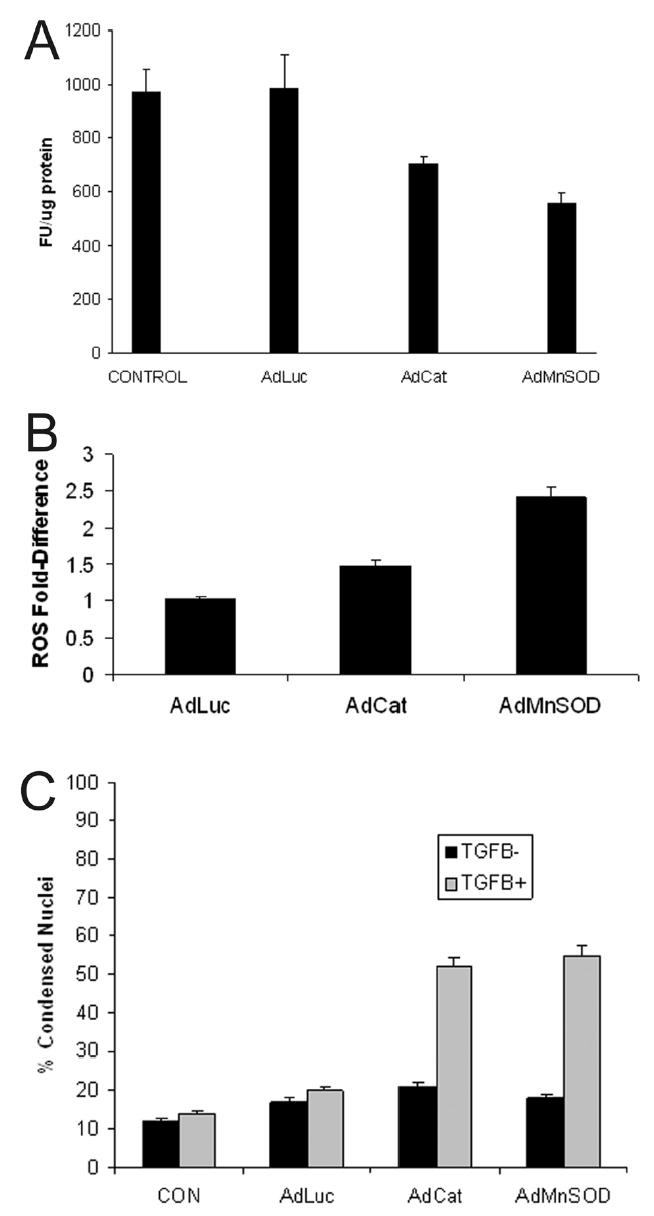
(A) Cirrhotic hepatocytes were transfected with adenoviruses (MOI 100) expressing luciferase (AdLuc; control), catalase (AdCat), or MnSOD (AdMnSOD) for 24 hours and ROS activity was fluorometrically determined using DCF. Some hepatocytes were untreated with adenovirus (control). Transfection with AdCat and AdMnSOD decreased significantly ROS formation (p<0.05 vs. control). (B) Cirrhotic hepatocytes were transfected with adenoviruses (MOI 100) expressing luciferase (AdLuc), catalase (AdCat), or MnSOD (AdMnSOD) 24 hours prior to TGFβ (5 ng/ml) treatment and incubated with DCF. Generation of ROS, expressed as fold-difference, was fluorometrically determined at 90 minutes following TGFβ administration. The adenoviruses expressing the antioxidants, catalase and MnSOD permitted a ROS burst at 90 minutes, indicating ROS responsiveness to TGFβ administration in cirrhotic hepatocytes. (C) The percent of condensed nuclei, indicative of morphologic apoptosis, was determined at 48 hours in cirrhotic hepatocytes in response to treatment with or without TGFβ (5 ng/ml) after pretreatment for 24 hours with adenoviruses (100 MOI) expressing luciferase (AdLuc), catalase (AdCat), or MnSOD (AdMnSOD). Transfection of cirrhotic hepatocytes with adenoviruses expressing the antioxidants, catalase and MnSOD, followed by treatment with TGFβ increased significantly the percent of apoptotic hepatocytes compared to control (p<0.05 vs. control).
To confirm that caspase-mediated apoptosis was the mode of cell death in cirrhotic hepatocytes, cell lysates from control and adenoviral infected cells were subjected to immunoblots for activated caspase-3. The primary antibody has affinity for the inactive, pre-cleaved, 35 kDa caspase-3 zymogen and the cleaved, activated, 17 kDa fragment. In Fig. 3A, cirrhotic hepatocytes transduced with the luciferase alone (AdLuc; lane 1) or luciferase followed by TGFβ (lane 2) did not exhibit the cleaved caspase-3 product. Likewise, infection with AdMnSOD alone (lane 3) failed to result in caspase-3 cleavage. However, when cells infected AdMnSOD for 24 hours were subsequently treated with TGFβ, caspase-3 cleavage occurred at 48 hours following TGFβ treatment (lane 4). Similar findings were evident for cirrhotic hepatocytes treated with AdCat (Fig. 3B). These immunoblot findings were substantiated by a caspase assay that showed only cirrhotic hepatocytes transduced with antioxidant enzymes and subsequently treated with TGFβ for 48 hours exhibited caspase-3 activation (Fig. 3C). Additionally, cirrhotic hepatocytes transduced with AdMnSOD and AdCat for 24 hours and pretreated for one hour with zVAD, a pan-caspase inhibitor, failed to undergo apoptosis in response to TGFβ, further confirming an apoptotic mode of cell death (Fig. 3D). Collectively, these data suggest that TGFβ-mediated cell death in cirrhotic hepatocytes pretreated with adenoviruses expressing antioxidants is caspase-dependent.
Figure 3.
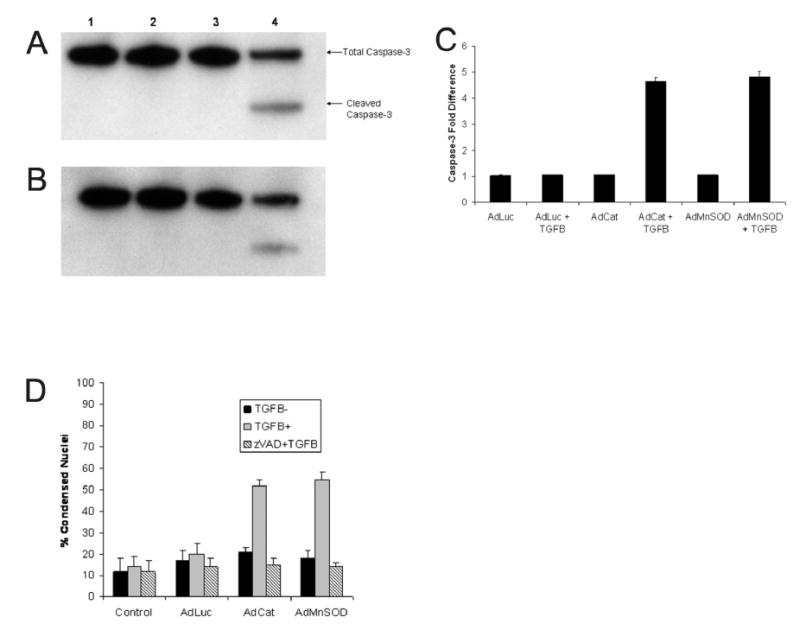
Cleavage of caspase-3 by immunoblot was assessed at 48 hours to confirm apoptotic cell death in cirrhotic hepatocytes transfected with adenoviruses (100 MOI) expressing luciferase (AdLuc), catalase (AdCat), or MnSOD (AdMnSOD) for 24 hours prior to treatment with TGFβ (5 ng/ml). (A) Cirrhotic hepatocytes were transfected with AdLuc alone (lane 1), AdLuc followed by TGFβ (lane 2), AdMnSOD alone (lane 3), or AdMnSOD followed by TGFβ (lane 4). As expected, only infection with AdMnSOD followed by TGFβ treatment resulted in the cleaved product indicating caspase-3 activity. (B) Cirrhotic hepatocytes were transfected with AdLuc alone (lane 1), AdLuc followed by TGFβ (lane 2), AdCat alone (lane 3), or AdCat followed by TGFβ (lane 4). The cleaved caspase-3 fragment was present only in AdCat infected hepatocytes treated with TGFβ. (C) Caspase-3 activity, expressed as fold-difference, was measured at 48 hours to confirm apoptotic cell death in cirrhotic hepatocytes transfected with adenviruses (100 MOI) expressing luciferase, catalase, or MnSOD for 24 hours prior to treatment with or without TGFβ (5 ng/ml). Infection with adenoviruses alone did not alter caspase-3 activity whereas transduction of cirrhotic hepatocytes with antioxidant expressing adenoviruses (AdCat and AdMnSOD) followed by TGFβ increased significantly caspase-3 activity. (D) The percent of condensed nuclei was determined 48 hours following TGFβ treatment in cirrhotic hepatocytes infected with adenoviruses (MOI 100) expressing luciferase (AdLuc), catalase (AdCat), or MnSOD (AdMnSOD) for 24 hours prior to TGFβ treatment (5 ng/ml). These hepatocytes were also pretreated with the pan-caspase inhibitor, zVAD (5 μM), for one hour prior to treatment with or without TGFβ. Pretreatment with zVAD inhibited apoptosis in cirrhotic hepatocytes infected with AdCat and AdMnSOD confirming a caspase-mediated form of cell death.
Co-localization of TGFβ-Induced ROS and Mitochondrial Function
Because AdMnSOD administration suggested a prominent role for mitochondria in ROS production, confocal microscopy was used to investigate TGFβ-induced ROS production in mitochondria. In these experiments, green-fluorescent H2-DCF (2 μM) was used to identify ROS and red fluorescent MTR (500 nM) was used to label mitochondria. The ROS response was monitored by confocal microscopy in both normal and cirrhotic hepatocytes that were untreated or pretreated with various antioxidants prior to administration of TGFβ. A significant increase in baseline ROS activity was evident in untreated cirrhotic hepatocytes compared to normal hepatocytes (Fig. 4A).
Figure 4.
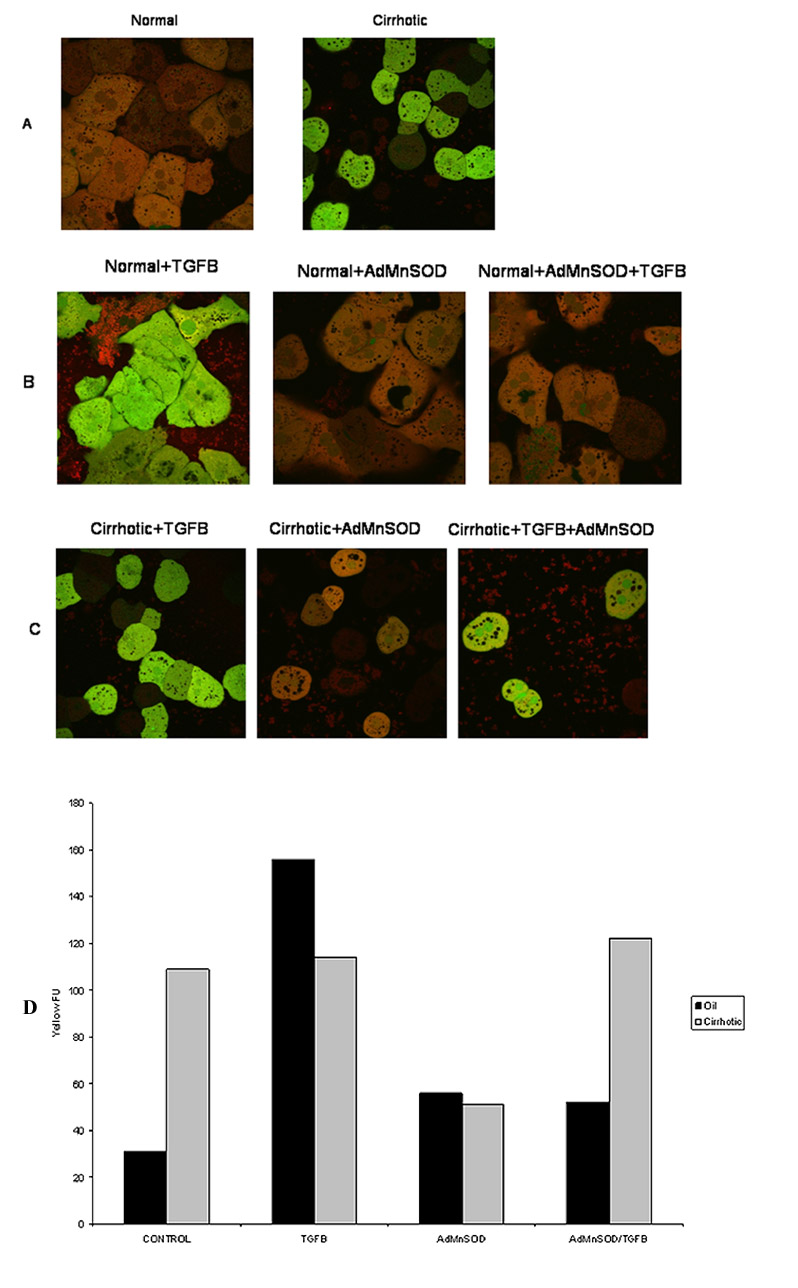
Normal and cirrhotic hepatocytes were loaded with the fluorophores H2-DCFDA (2 μM; green) to assess ROS formation and MitoTracker Red (MTR 500 nM; red) to localize respiring mitochondria. Confocal microscopy was performed at 90 minutes following TGFβ (5 ng/ml) administration. (A) Normal (left) and cirrhotic (right) hepatocytes were imaged without treatment. Note a low mitochondrial ROS formation in normal hepatocytes while diffused ROS formation in cirrhotic hepatocytes. (B) Normal hepatocytes were treated with TGFβ for 90 min. Some hepatocytes were transfected with AdMnSOD for 24 hours prior to TGFβ treatment. Confocal imaging revealed a ROS burst in the mitochondria (yellow fluorescence, left panel), which was suppressed by AdMnSOD transfection (middle and right panel). (C) Confocal imaging of cirrhotic hepatocytes. TGFβ alone did not caused a mitochondrial ROS burst (left panel). Although AdMnSOD alone (middle panel) decreased ROS generation, subsequent TGFβ administration induced a mitochondrial ROS burst (yellow, right panel). (D) Quantification of yellow fluorescence, an indication of mitochondrial generation of ROS for control and cirrhotic hepatocytes treated with TGFβ, AdMnSOD or TGFβ and AdMnSOD.
Similar to our previous study [1], normal hepatocytes treated with TGFβ underwent a ROS burst at 90 min (Fig. 4B); however, when these normal hepatocytes were pretreated with AdMnSOD and then administered TGFβ, a ROS burst did not occur. In normal hepatocytes, ROS levels did not change significantly in response to antioxidant treatment alone. Similar results were noted when AdCat and trolox were used as antioxidants.
In cirrhotic hepatocytes, AdMnSOD infection decreased significantly the basal ROS activity (Fig. 4C). Cirrhotic hepatocytes treated with TGFβ alone did not undergo a ROS spike at 90 min, but cirrhotic cells pretreated with AdMnSOD underwent a ROS burst similar to that of untreated control cells (Fig. 4D).
These data confirm that a reduction in ROS levels in cirrhotic hepatocytes sensitizes these previously resistant cells to TGFβ-induced mitochondrial ROS generation and subsequent apoptosis. Conversely, treatment of normal hepatocytes with various antioxidants renders these formerly responsive hepatocytes resistant to TGFβ-induced mitochondrial ROS generation.
Role of Anti-Apoptotic Proteins and Protein Tyrosine Phosphatase in Cirrhotic Livers and Hepatocytes
To test if resistance to apoptosis in cirrhotic hepatocytes could result from modified apoptotic machinery, lysates from normal and cirrhotic livers were immunoblotted for anti-apoptotic proteins, including Bcl-xL and MCL-1 [12]. Levels of both Bcl-xL and MCL-1 from cirrhotic livers were similar to those from normal livers (Fig. 5A), suggesting that upregulation of anti-apoptotic proteins is not the mechanism underlying resistance to apoptosis in cirrhotic livers.
Figure 5.
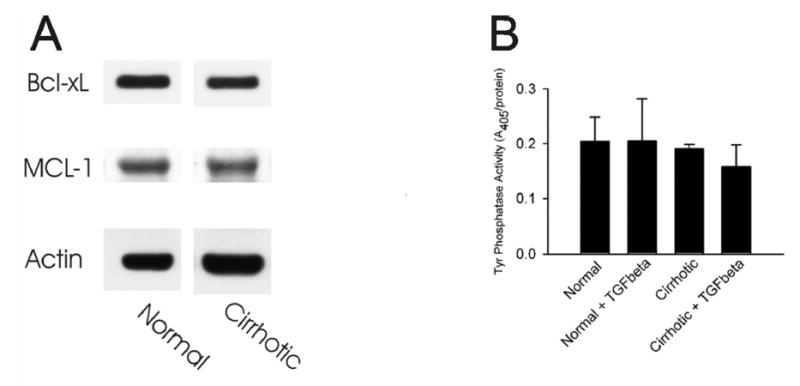
(A) Whole tissue lysates were prepared from normal and cirrhotic livers and levels of Bcl-xL and MCL-1 were determined by Western blot analysis. Actin levels were also immunoblotted to ensure an equal protein loading. (B) Hepatocytes isolated from normal and cirrhotic livers were incubated in HDM in the presence or absence of 5 ng/ml TGFβ, as described in MATERIALS and METHODS. After 48 hours, cell lysates were prepared and total protein tyrosine phosphatase activity was determined spectrometrically.
ROS can modify proteins, lipids and nucleic acids, leading to tissue injury. Since protein tyrosine phosphatase is a key signaling molecule associated with TGFβ transduction and is also known to be sensitive to oxidative stress [13,14], we examined the possibility that enhanced basal ROS in cirrhotic hepatocytes could perturb the activity of protein tyrosine phosphatase. Measurement of total tyrosine phosphatase activity demonstrates that there was no significant difference in tyrosine phosphatase activity between normal and cirrhotic hepatocytes (Fig. 5B). Moreover, treatment with TGFβ for 48 hours did not change the activity of tyrosine phosphatase in both groups (Fig. 5B). These findings suggest that protein tyrosine phosphatases are not significantly involved in cirrhotic hepatocyte resistance to apoptosis, and the mechanisms other than altered tyrosine phosphatase may contribute to resistance to apoptosis.
H2O2-Treated Normal Hepatocytes Mimic Cirrhotic Hepatocyte ROS Activity and Apoptotic Response
Our previous work [1] and the findings in this study suggest strongly that basal ROS activity in cirrhotic hepatocytes mediates responsiveness to TGFβ-induced apoptosis. To determine if normal hepatocytes, which are responsive to TGFβ-induced apoptosis, could be rendered resistant to apoptosis by increased basal ROS activity, these cells were exposed to H2O2 and ROS activity monitored over 72 hours. Treatment with 10 μM H2O2 for produced sustained ROS activity comparable to cirrhotic hepatocyte basal ROS activity (Fig. 6A). Additionally, hepatocyte viability was 97% over 72 hours, indicating that this low concentration of H2O2 is not cytotoxic (data not shown). When these H2O2-converted hepatocytes were subsequently treated with TGFβ, an ROS burst was not apparent (data not shown). Furthermore, these H2O2-converted hepatocytes were resistant to TGFβ-induced apoptosis (Fig. 6B). Trolox pretreatment of normal hepatocytes resulted in the expected inhibitory response to TGFβ-induced apoptosis compared to normal hepatocytes not treated with antioxidant. However, trolox pretreatment of H2O2-converted hepatocytes resulted in a decrease in ROS activity and return of the TGFβ-induced apoptotic response. To further evaluate the role of enhanced ROS in cirrhotic hepatocytes, normal hepatocytes were treated with 30 μM 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), an intracellular redox-cycling agent [15], for 20 min. After washing once, hepatocytes were further incubated in HDM (without DMNQ) for 24 hours in the presence of TGFβ. Development of apoptosis was assessed by chromatin condensation and nuclear fragmentation in PI-stained nuclei, as described in MATERIALS and METHODS (Fig. 7). DMNQ at this concentration was not cytotoxic (data not shown). In normal hepatocytes, TGFβ treatment substantially increased apoptosis (Fig. 7A). In contrast, DMNQ-converted hepatocytes were resistant to TGFβ-mediated apoptosis (Fig. 7B). Taken together, these findings confirm the importance of the basal ROS activity in preventing TGFβ-induced hepatocyte apoptosis.
Figure 6.
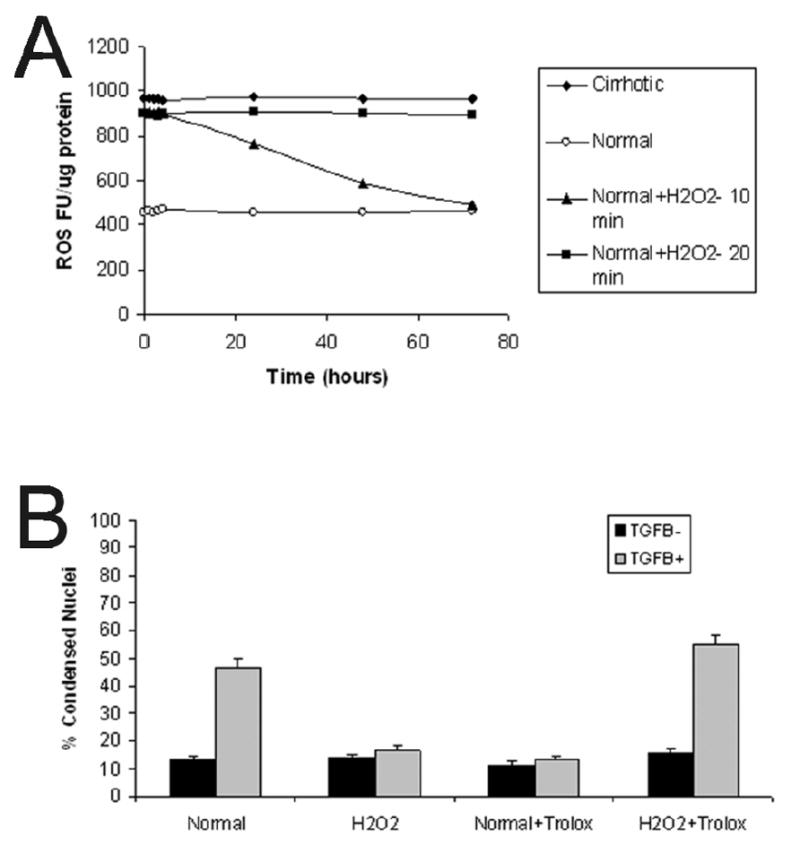
(A) ROS fluorescent units (FU) were determined over time in normal, cirrhotic, and normal-H2O2-converted hepatocytes. Normal hepatocytes (open circles) have a low basal level of ROS activity whereas cirrhotic hepatocytes (black diamonds) have a high steady-state level of ROS activity. Normal hepatocytes exposed to 10 μM H2O2 for 10 minutes (black triangles) failed to maintain a sustained ROS response. However, normal hepatocytes exposed to 10 μM H2O2 for 20 minutes (black squares) demonstrate sustained increased ROS activity similar to basal cirrhotic hepatocytes over the study period. These hepatocytes had 97% viability (data not shown). (B) The percent of condensed nuclei was determined in normal and H2O2-converted hepatocytes at 48 hours after pretreatment (or not) with trolox (2 μM) followed by treatment with or without TGFβ (5 ng/ml). Treatment with H2O2 alone did not increase apoptosis, and, similar to cirrhotic hepatocytes, H2O2-converted cells were resistant to TGFβ -induced apoptosis at 48 hours. However, H2O2-converted hepatocytes that were pretreated with trolox prior to TGFβ exposure underwent apoptosis similar to normal hepatocyte controls and antioxidant-treated cirrhotic hepatocytes.
Figure 7.
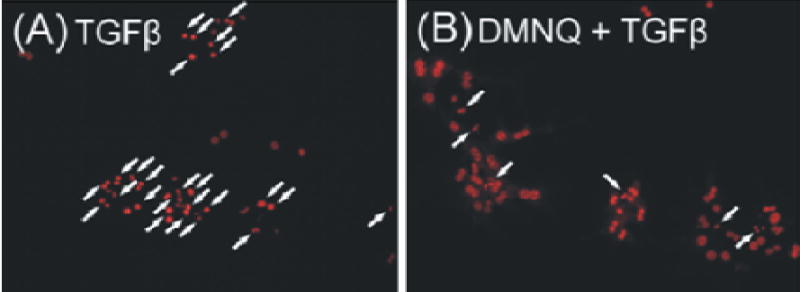
Hepatocytes isolated from normal livers were incubated with 5 ng/ml TGFβ for 24 h. Some hepatocytes were treated with 30 μM 2,3-dimethoxy-1,4-naphthoquinone (DMNQ) for 20 min prior to TGFβ administration. After washing once, hepatocytes were further incubated with TGFβ. Apoptosis was evaluated by chromatin condensation and nuclear fragmentation of PI-stained nuclei (arrows), as described in MATERIALS and METHODS. TGFβ alone induced a substantial apoptosis (A), which was reversed by a brief treatment with DMNQ (B).
DISCUSSION
ROS serve as intermediaries in cellular signaling pathways and recent studies have demonstrated the importance of generating these molecules in apoptotic death pathways [1,4,16]. Our previous work examining cirrhotic hepatocytes suggests that chronic elevation of basal ROS activity inhibits mitochondrial pathway-dependent hepatocyte apoptosis induced by TGFβ, TNFα and UV, and the findings in this study document the crucial role for ROS in TGFβ-induced hepatocyte apoptosis. The aim of this study was to investigate further the necessity of ROS in hepatocyte apoptosis, and specifically to examine the importance of mitochondrial-generated ROS activity. The major findings of this study corroborate our previous work and extend our findings to suggest that the basal increase in cirrhotic hepatocyte ROS likely is not limited to a single ROS generating pathway. Furthermore, we found that the mitochondria are a primary source of TGFβ-induced ROS activity in normal hepatocytes and in cirrhotic hepatocytes that have been pretreated with antioxidants. Finally, normal hepatocytes exposed to H2O2 and DMNQ can be “converted” to an oxidative state that confers resistance to TGFβ-induced apoptosis. Cumulatively, these findings suggest that ROS play an integral role in hepatocyte responsiveness to apoptotic stimuli.
The cirrhotic liver represents the end-stage morphologic result from chronic inflammatory injury related to viral hepatitis, alcohol ingestion, and other metabolic causes. Chronic CCl4-induced liver injury in the mouse produces bridging fibrosis with up-regulation of fibrotic stimuli such as TGFβ [17]. Despite increased hepatic levels of TGFβ and other pro-apoptotic cytokines, the cirrhotic liver does not have a chronically increased rate of apoptosis [1], which suggests that the hepatocyte exposed to chronic inflammation has adapted an anti-apoptotic mechanism. The anti-apoptotic phenotype has been noted in several forms of liver injury including alcohol-induced liver injury which is associated with increased ROS [18]. In that study, hepatocellular apoptosis was decreased with a resultant increase in dysplastic hepatocytes and these phenotypic changes were likely related to p53 expression. Furthermore, increased ROS activity has been linked to malignant transformation [19] which may be related to cytokine receptor profiles and resistance to apoptosis [20] or to decreased expression of antioxidants [8,21]. In addition to decreasing responsiveness to apoptosis, increased ROS may result in negative regulatory changes in the cell cycle and alter the balance between proliferation and apoptosis [22]. Collectively, these findings suggest that chronically increased ROS may mediate changes in susceptibility to apoptosis and as demonstrated by our findings and those of Herrera et al. [4,23,24] multiple ROS generating pathways and downstream cellular signaling pathways may be involved.
Because the mitochondria are a primary source of ROS, we [1] and others [25] have focused on the importance of mitochondrial ROS generation in chronic hepatic inflammatory states. The current study demonstrates that TGFβ-induced ROS activity in normal hepatocytes predominantly arises from the mitochondria as evidenced by co-localization of H2-DCFDA fluorescent activity and respiring mitochondria on confocal microscopic imaging. Moreover, in cirrhotic hepatocytes that are pretreated with an antioxidant and subsequently treated with TGFβ, significant ROS activity again arises in the mitochondria suggesting that the mitochondria are an important source of TGFβ-induced ROS. These findings also suggest that even though cirrhotic hepatocytes have elevated basal ROS activity, the mitochondria in these cells are functionally intact, respond appropriately after treatment with an antioxidant, and do not have an irreversible injury related to CCl4 administration. The mechanisms that may influence mitochondrial ROS production have been examined closely and may be related to the Bcl-family of proteins [26–28]. However, our results suggest that up-regulation of anti-apoptotic proteins, Bcl-xL and MCL-1, is not the mechanism underlying resistance to apoptosis in cirrhotic hepatocytes since levels of both proteins in cirrhotic livers were similar to those in normal livers. In receptor-dependent apoptosis mitochondrial ROS production may be related to a Bid-mediated induction of mitochondrial ROS [27]. Alternatively, Bax, another pro-apoptotic Bcl-family member, may associate with the mitochondrial voltage-dependent anion channel and modulate cytochrome c release and apoptotic cell death in the acute phase [28]. In the setting of chronic inflammation, mitochondrial function and Bcl-family function have been less studied, but in nonalcoholic steatohepatitis (NASH) changes in the P450 system may up-regulate ROS production [25]. These studies suggest that several pathways may be involved in mitochondrial ROS generation, and that further investigation is needed to determine how these pathways may mediate ROS production in chronic inflammatory states.
Our experiments in which normal murine hepatocytes were exposed to H2O2 and exhibited sustained ROS activity and antioxidant-reversible resistance to apoptosis suggests that the redox state of hepatocytes mediates the TGFβ-apoptotic response. The importance of redox state is further supported by our findings that a brief treatment of DMNQ, a redox-cycling agent [15], to normal hepatocytes reversed the sensitivity to TGFβ-induced apoptosis. Similar findings were noted by Tejima et al. [29] who used H2O2 as a preconditioning molecule and demonstrated the hepatotoxicity could be reduced by low-dose H2O2. Likewise, hepatocytes treated with nontoxic doses of menadione, a superoxide generator, resisted oxidant-induced cell death through an ERK-dependent pathway while the JNK pathway was pro-apoptotic [30]. Interestingly, the pro-apoptotic function of the JNK pathway appeared to be mitochondrial-independent. Other mechanisms that may render hepatocytes resistant to H2O2-mediated cell death involve intramitochondrial changes in caspase processing [31]. In this study, cells treated with H2O2 demonstrated a slight decrease in the inner mitochondrial membrane potential but without cytochrome c release. In addition, within the mitochondria, procaspase-9 underwent autocleavage, but overexpression of the antiapoptotic protein, Bcl-2, caused accumulation of caspase-9 and prevented oxidant-induced cell death. The intramitochondrial processing of procaspase-9 was associated with disulfide-bonded dimers of caspase-9 that appeared to prevent mitochondrial release. These studies suggest that exposure to nontoxic doses of oxidants can prevent cell death by up-regulation of anti-apoptotic cellular mechanisms.
In conclusion, these experiments demonstrate that the ROS state of hepatocytes mediates responsiveness to TGFβ-induced apoptosis. In normal hepatocytes, which have low basal ROS levels, a mitochondrial-derived ROS burst is required for TGFβ induced apoptosis whereas in cirrhotic hepatocytes, which have an increased basal level of ROS, pretreatment with an antioxidant permits a TGFβ-induced mitochondrial ROS response with subsequent apoptosis. These studies further suggest that the mitochondria in cirrhotic hepatocytes are not irreversibly damaged and that adequate energy stores are present to permit apoptosis. The mechanisms that regulate these ROS-mediated responses are unknown and warrant further investigation.
References
- 1.Black D, Bird MA, Samson CM, Lyman S, Lange PA, Schrum LW, Qian T, Lemasters JJ, Brenner DA, Rippe RA, Behrns KE. Primary cirrhotic hepatocytes resist TGFbeta-induced apoptosis through a ROS-dependent mechanism. J Hepatol. 2004;40:942–951. doi: 10.1016/j.jhep.2004.02.031. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez A, Alvarez AM, Benito M, Fabregat I. Apoptosis induced by transforming growth factor-beta in fetal hepatocyte primary cultures: involvement of reactive oxygen intermediates. J Biol Chem. 1996;271:7416–7422. doi: 10.1074/jbc.271.13.7416. [DOI] [PubMed] [Google Scholar]
- 3.Herrera B, Alvarez AM, Sanchez A, Fernandez M, Roncero C, Benito M, Fabregat I. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor (beta) in fetal hepatocytes. FASEB J. 2001;15:741–751. doi: 10.1096/fj.00-0267com. [DOI] [PubMed] [Google Scholar]
- 4.Herrera B, Murillo MM, Alvarez-Barrientos A, Beltran J, Fernandez M, Fabregat I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic Biol Med. 2004;36:16–26. doi: 10.1016/j.freeradbiomed.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 5.Albright CD, Salganik RI, Craciunescu CN, Mar MH, Zeisel SH. Mitochondrial and microsomal derived reactive oxygen species mediate apoptosis induced by transforming growth factor-beta1 in immortalized rat hepatocytes. J Cell Biochem. 2003;89:254–261. doi: 10.1002/jcb.10498. [DOI] [PubMed] [Google Scholar]
- 6.Franklin CC, Rosenfeld-Franklin ME, White C, Kavanagh TJ, Fausto N. TGFbeta1-induced suppression of glutathione antioxidant defenses in hepatocytes: caspase-dependent post-translational and caspase-independent transcriptional regulatory mechanisms. FASEB J. 2003;17:1535–1537. doi: 10.1096/fj.02-0867fje. [DOI] [PubMed] [Google Scholar]
- 7.Cullen JJ, Hinkhouse MM, Grady M, Gaut AW, Liu J, Zhang YP, Weydert CJ, Domann FE, Oberley LW. Dicumarol inhibition of NADPH:quinone oxidoreductase induces growth inhibition of pancreatic cancer via a superoxide-mediated mechanism. Cancer Res. 2003;63:5513–5520. [PubMed] [Google Scholar]
- 8.Weydert C, Roling B, Liu J, Hinkhouse MM, Ritchie JM, Oberley LW, Cullen JJ. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol Cancer Ther. 2003;2:361–369. [PubMed] [Google Scholar]
- 9.Buckman JF, Hernandez H, Kress GJ, Votyakova TV, Pal S, Reynolds IJ. MitoTracker labeling in primary neuronal and astrocytic cultures: influence of mitochondrial membrane potential and oxidants. J Neurosci Methods. 2001;104:165–176. doi: 10.1016/s0165-0270(00)00340-x. [DOI] [PubMed] [Google Scholar]
- 10.Elmore SP, Nishimura Y, Qian T, Herman B, Lemasters JJ. Discrimination of depolarized from polarized mitochondria by confocal fluorescence resonance energy transfer. Arch Biochem Biophys. 2004;422:145–152. doi: 10.1016/j.abb.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 11.Montalibet J, Skorey KI, Kennedy BP. Protein tyrosine phosphatase: enzymatic assays. Methods. 2005;35:2–8. doi: 10.1016/j.ymeth.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 12.Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37:267–271. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 14.Greenwel P, Hu W, Kohanski RA, Ramirez F. Tyrosine dephosphorylation of nuclear proteins mimics transforming growth factor beta 1 stimulation of alpha 2(I) collagen gene expression. Mol Cell Biol. 1995;15:6813–6819. doi: 10.1128/mcb.15.12.6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dypbukt JM, Ankarcrona M, Burkitt M, Sjoholm A, Strom K, Orrenius S, Nicotera P. Different prooxidant levels stimulate growth, trigger apoptosis, or produce necrosis of insulin-secreting RINm5F cells. The role of intracellular polyamines. J Biol Chem. 1994;269:30553–30560. [PubMed] [Google Scholar]
- 16.Ribeiro A, Bronk SF, Roberts PJ, Urrutia R, Gores GJ. The transforming growth factor beta(1)-inducible transcription factor TIEG1, mediates apoptosis through oxidative stress. Hepatology. 1999;30:1490–1497. doi: 10.1002/hep.510300620. [DOI] [PubMed] [Google Scholar]
- 17.Tsukada S, Westwick JK, Ikejima K, Sato N, Rippe RA. SMAD and p38 MAPK signaling pathways independently regulate alpha1(I) collagen gene expression in unstimulated and transforming growth factor-beta-stimulated hepatic stellate cells. J Biol Chem. 2005;280:10055–10064. doi: 10.1074/jbc.M409381200. [DOI] [PubMed] [Google Scholar]
- 18.Pani G, Fusco S, Colavitti R, Borrello S, Maggiano N, Cravero AA, Farre SM, Galeotti T, Koch OR. Abrogation of hepatocyte apoptosis and early appearance of liver dysplasia in ethanol-fed p53-deficient mice. Biochem Biophys Res Commun. 2004;325:97–100. doi: 10.1016/j.bbrc.2004.09.213. [DOI] [PubMed] [Google Scholar]
- 19.O’Byrne KJ, Dalgleish AG. Chronic immune activation and inflammation as the cause of malignancy. Br J Cancer. 2001;85:473–483. doi: 10.1054/bjoc.2001.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon DY, Cho YS, Park JW, Kim SH, Kim JW. Up-regulation of reactive oxygen species (ROS) and resistance to Fas-mediated apoptosis in the C33A cervical cancer cell line transfected with IL-18 receptor. Clin Chem Lab Med. 2004;42:499–506. doi: 10.1515/CCLM.2004.085. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Hinkhouse MM, Sun W, Weydert CJ, Ritchie JM, Oberley LW, Cullen JJ. Redox regulation of pancreatic cancer cell growth: role of glutathione peroxidase in the suppression of the malignant phenotype. Hum Gene Ther. 2004;15:239–250. doi: 10.1089/104303404322886093. [DOI] [PubMed] [Google Scholar]
- 22.Horimoto M, Fulop P, Derdak Z, Wands JR, Baffy G. Uncoupling protein-2 deficiency promotes oxidant stress and delays liver regeneration in mice. Hepatology. 2004;39:386–392. doi: 10.1002/hep.20047. [DOI] [PubMed] [Google Scholar]
- 23.Herrera B, Fernandez M, Alvarez AM, Roncero C, Benito M, Gil J, Fabregat I. Activation of caspases occurs downstream from radical oxygen species production, Bcl-xL down-regulation, and early cytochrome C release in apoptosis induced by transforming growth factor beta in rat fetal hepatocytes. Hepatology. 2001;34:548–556. doi: 10.1053/jhep.2001.27447. [DOI] [PubMed] [Google Scholar]
- 24.Herrera B, Fernandez M, Roncero C, Ventura JJ, Porras A, Valladares A, Benito M, Fabregat I. Activation of p38MAPK by TGF-beta in fetal rat hepatocytes requires radical oxygen production, but is dispensable for cell death. FEBS Lett. 2001;499:225–229. doi: 10.1016/s0014-5793(01)02554-6. [DOI] [PubMed] [Google Scholar]
- 25.Fromenty B, Robin MA, Igoudjil A, Mansouri A, Pessayre D. The ins and outs of mitochondrial dysfunction in NASH. Diabetes Metab. 2004;30:121–138. doi: 10.1016/s1262-3636(07)70098-8. [DOI] [PubMed] [Google Scholar]
- 26.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000;6:513–519. doi: 10.1038/74994. [DOI] [PubMed] [Google Scholar]
- 27.Ding WX, Ni HM, DiFrancesca D, Stolz DB, Yin XM. Bid-dependent generation of oxygen radicals promotes death receptor activation-induced apoptosis in murine hepatocytes. Hepatology. 2004;40:403–413. doi: 10.1002/hep.20310. [DOI] [PubMed] [Google Scholar]
- 28.Adachi M, Higuchi H, Miura S, Azuma T, Inokuchi S, Saito H, Kato S, Ishii H. Bax interacts with the voltage-dependent anion channel and mediates ethanol-induced apoptosis in rat hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2004;287:G695–G705. doi: 10.1152/ajpgi.00415.2003. [DOI] [PubMed] [Google Scholar]
- 29.Tejima K, Arai M, Ikeda H, Tomiya T, Yanase M, Inoue Y, Nagashima K, Nishikawa T, Watanabe N, Omata M, Fujiwara K. Ischemic preconditioning protects hepatocytes via reactive oxygen species derived from Kupffer cells in rats. Gastroenterology. 2004;127:1488–1496. doi: 10.1053/j.gastro.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 30.Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology. 2003;37:1405–1413. doi: 10.1053/jhep.2003.50233. [DOI] [PubMed] [Google Scholar]
- 31.Katoh I, Tomimori Y, Ikawa Y, Kurata S. Dimerization and processing of procaspase-9 by redox stress in mitochondria. J Biol Chem. 2004;279:15515–15523. doi: 10.1074/jbc.M311819200. [DOI] [PubMed] [Google Scholar]