

Summary
In a recent clinical trial involving patients with metastatic melanoma, immunosuppressive conditioning with fludarabine and cyclophosphamide resulted in a 50% response rate and a robust long-term persistence of adoptively transferred T cells. Experimental findings indicate that lymphodepletion prior to adoptive transfer of tumor-specific T lymphocytes plays a key role in enhancing treatment efficacy by eliminating regulatory T cells and competing elements of the immune system (‘cytokine sinks’). Newly emerging animal data suggest that more profound lymphoablative conditioning with autologous hematopoetic stem-cell rescue might further enhance treatment results. Here we review recent advances in adoptive immunotherapy of solid tumors and discuss the rationale for lymphodepleting conditioning. We also address safety issues associated with translating experimental animal results of total lymphoid ablation into clinical practice.
Review criteria
The PubMed and MEDLINE databases were searched for articles published until April 2006. Electronic early-release publications were also included. Only articles published in English were considered. The search terms used included “adoptive cell transfer”, “lymphodepletion”, “lymphopenia”, “TBI”, “homeostatic proliferation”, “allogeneic transplant”, “syngeneic transplant”, “IPS” and “treatment related mortality”. Full articles were obtained and references were checked for additional material when appropriate. References were chosen based on the best clinical or laboratory evidence, especially if data had been corroborated by published work from other centers. Priority was given to studies in high-impact-factor journals when available.
Keywords: adoptive cell transfer, immunodepletion, lymphodepletion, melanoma, T lymphocytes Top of page
Introduction
Experiments in both mice and humans have established that the immune system can damage and in some cases destroy even very large established tumors. Treatment with interleukin (IL-)2 has been effective in approximately 10–20% of patients with melanoma1, 2, 3, 4 or renal cell carcinoma.2 Unfortunately, clinical trials of tumor-specific active immunotherapy (i.e. vaccination) have so far been disappointing, with objective response rates of 5% or less regardless of histology.5 Effective therapeutic vaccination has been limited by many factors, including absence or low frequency and low avidity of antigen-specific T-cell precursors directed against self-antigens as a result of thymic negative selection,6, 7 and various peripheral inhibitory mechanisms.7, 8, 9, 10 Tumor outgrowth is clearly not prevented even when large numbers of specific T cells are generated in high-risk cancer patients without clinical or radiological evidence of disease at the initiation of immunization.11 At the same time, it has become evident that the use of immunotherapy based on adoptive cell transfer (ACT) can overcome many of the hurdles that hinder current vaccine-based approaches. Using ACT, large numbers of in vitro expanded and activated tumor-specific T cells can be delivered rapidly into the patient’s immune system.12, 13 Unfortunately, multiple factors, including tumor immune evasion, homeostasis and induction of tolerance, as well as suboptimal quality of transferred T cells, have hampered the otherwise promising attempts at driving tumor rejection.14, 15, 16
Recently a 50% response rate according to RECIST criteria was reported in patients with metastatic melanoma treated with in vitro expanded tumor-infiltrating lymphocytes (TILs) and IL-2 following a lymphodepleting nonmyeloablative preparative regimen of cyclophosphamide and fludarabine.17, 18 This significant achievement was attributed to the key realization that the host’s immune system needs to be properly conditioned, in order to create an appropriate ‘lymphoid space’ that is devoid of regulatory mechanisms. Thus, lymphodepletion enables the host to accommodate transferred T lymphocytes and gives these cells an advantage over other competing cellular populations.19, 20
Immunodepletion and ACT have not yet been tested in randomized studies, but the results of recent clinical trials using these methods are notable for their unprecedented response rates and the fact that the patients studied had previously failed other modes of immunotherapy including high-dose IL-2 and ACT;17 however, success of the current approaches is primarily limited to patients with melanoma and we are still far from offering a cure for the majority of patients. More effort needs to be aimed at developing treatment regimens that are reliably effective, easily implemented and accessible, and yet maintain acceptable safety and toxicity profiles. Here we discuss recent advances in clinical applications of adoptive immune therapy, including the theoretical and preclinical bases of lymphodepleting conditioning. We hypothesize, based on animal data, that causing more immune ablation in patients could potentially lead to the further augmentation of the antitumor effect. We will also discuss the safety issues related to the development of this new strategy, applying experience from the related field of allogeneic and autologous stem-cell transplantation.
Adoptive cell transfer therapy—recent clinical experiences
ACT has been conceived as a means of providing high numbers of tumor-specific T cells that are expanded and activated in vitro. In some earlier trials, CD8+ T-cell clones specific for the melanoma antigens MART-1 and gp100 could be consistently generated from patients, but the T-cell clones declined rapidly after ACT and the patients failed to produce meaningful clinical response.15, 16 Another approach was to use TILs obtained from surgically resected tumors, which were activated and expanded in vitro utilizing allogeneic feeder cells, OKT-3 monoclonal antibody and IL-2.21 These cultured cells were highly reactive against human leukocyte antigen (HLA) A2 melanoma cell lines and autologous tumor, but demonstrated only moderate success after ACT and treatment with high-dose IL-2 (720,000 IU/kg every 8 h; overall response rate 34%). A preconditioning regimen of cyclophosphamide (25 mg/kg) alone did not measurably add to the overall response rate.
On the basis of animal data, it has been postulated that lymphodepletion may enhance the effectiveness of adoptively transferred T cells, and that a manipulation of the recipient immune environment might improve the treatment outcome.22 This hypothesis has been clinically tested in the setting of hematologic malignancy in a series of heavily pretreated patients with refractory non-Hodgkin lymphoma, who received infusions of autologous lymphocytes ex vivo cultured with anti-CD3 and anti-CD28 following high-dose chemotherapy and autologous CD34+ stem-cell transplant. Rapid recovery of lymphocyte compartment was observed and in some cases significant delayed lymphocytosis occurred.20 A similar approach has been utilized in patients with metastatic melanoma refractory to conventional treatments.17 The patients received a highly lymphodepleting conditioning regimen consisting of cyclophosphamide (60 mg/kg for 2 days) and fludarabine (25 mg/m2 for 5 days) before adoptive transfer of TILs. Patients were subsequently treated with high-dose IL-2. Some patients also received a second infusion of T cells. Objective responses according to RECIST criteria23 were seen in 6 out of 13 patients treated (47%), with mixed responses with decrease at only some sites of the disease in 4 additional patients. The majority of responders demonstrated antimelanocyte autoimmunity, including vitiligo and uveitis. Molecular analysis and in vitro functional data showed for the first time that some patients developed striking post-transfer clonal lymphocytosis (up to 21,000 cells/ μl on day 7 in one case) with robust long-term persistence and significant skewing of the T-cell repertoire towards the adoptively infused tumor-reactive population.16 These findings were in sharp contrast to previously published highly discouraging observations of persistence of adoptively transferred T cells, which were undetectable by polymerase chain reaction after only several hours or days.16, 24
A follow-up study to the same protocol was published 2 years later.25 This trial included a total of 35 patients with an overall objective response of 51%—3 complete responses and 15 partial responses. As expected, observed toxicities were due to high-dose IL-2 infusions and myelosuppression. All patients recovered from these initial toxicities with supportive care. One patient died of Epstein–Barr virus (EBV) lymphoproliferative disease. The nadir of peripheral lymphocyte count occurred at a median of 17 cells/ μl on the day of transfer. In some patients transient lymphocytosis was seen within the first week after transfer, even before the earliest evidence of monocyte recovery. The CD4+ T-cell counts were significantly diminished for a minimum of 1 year after treatment, while numbers of CD8+ cells were slightly increased and gradually normalized.25
Rationale for immunodepletion
The notion that the lymphopenic state enhances the efficacy of adoptively transferred T cells has been known for more than 20 years, although for a long time the mechanisms were not clear. In the early 1980s it was demonstrated that ACT of tumor-sensitized lymphocytes was effective only if the recipient was T-cell-deficient by thymectomy and irradiation.26 In another model, CD8+ T cells isolated from tumor-draining lymph nodes of mice bearing MCA 205 sarcoma actively proliferated and rejected the pulmonary metastases after total body irradiation (TBI).27
We have explored the role of lymphodepletion using a transgenic mouse model expressing the pmel-1 T-cell receptor (TCR), which is specific for the murine gp100 melanoma-associated antigen.28 Surprisingly, these mice are not protected against growth of B16 melanoma in spite of a very high frequency of tumor-specific CD8+ T cells (>90%). Treatment of tumor-bearing C57B6 mice is possible with adoptive transfer of pmel-1 T cells, but successful therapy also requires administration of IL-2 and effective vaccination with an altered (human gp100) peptide ligand vaccine.28 Gattinoni et al. have recently shown the striking effect of lymphodepletion on the utility of ACT in this model.21 Even though a response to treatment was observed in nonirradiated recipients, improvement of the therapy in sublethally irradiated (5 Gy) recipients was highly significant.
Multiple mechanisms explain the above observations (Figure 1). The size of the lymphoid compartment is tightly controlled by homeostatic factors, including access to antigen-presenting cells (APCs) and major histocompatibility complex (MHC) and self and antigenic peptides, and direct influence of different host T lymphocytes including regulatory cells CD4+CD25+FOXP3+Treg, as well as the competition for cytokines such as IL-2, IL-7, IL-15 and IL-21.29, 30, 31, 32, 33, 34, 35, 36 T cells, particularly those of a naive phenotype, undergo a process called homeostatic expansion (lymphopenia-induced homeostatic proliferation) after transfer into the lymphopenic host. This expansion is thought to be driven by homeostatic cytokines and exposure to self-peptides and other antigens. During this process, naive T cells change their phenotype into activated/effector cells (e.g. CD44high, CD62Llow and CD122high) and acquire effector abilities, as manifested by in vitro production of interferon-γ and cytolytic function.37, 38, 39, 40 This activation of T cells driven by homeostatic factors might lead to autoimmunity in a lymphopenic host (e.g. development of colitis41 or diabetes mellitus in nonobese diabetic mice42) or paradoxically promote graft rejection.43 Similarly, it has been shown that CD8+ T cells can acquire antitumor characteristics when transferred into immunodeficient mice.44
Figure 1. The rationale for lymphodepletion.
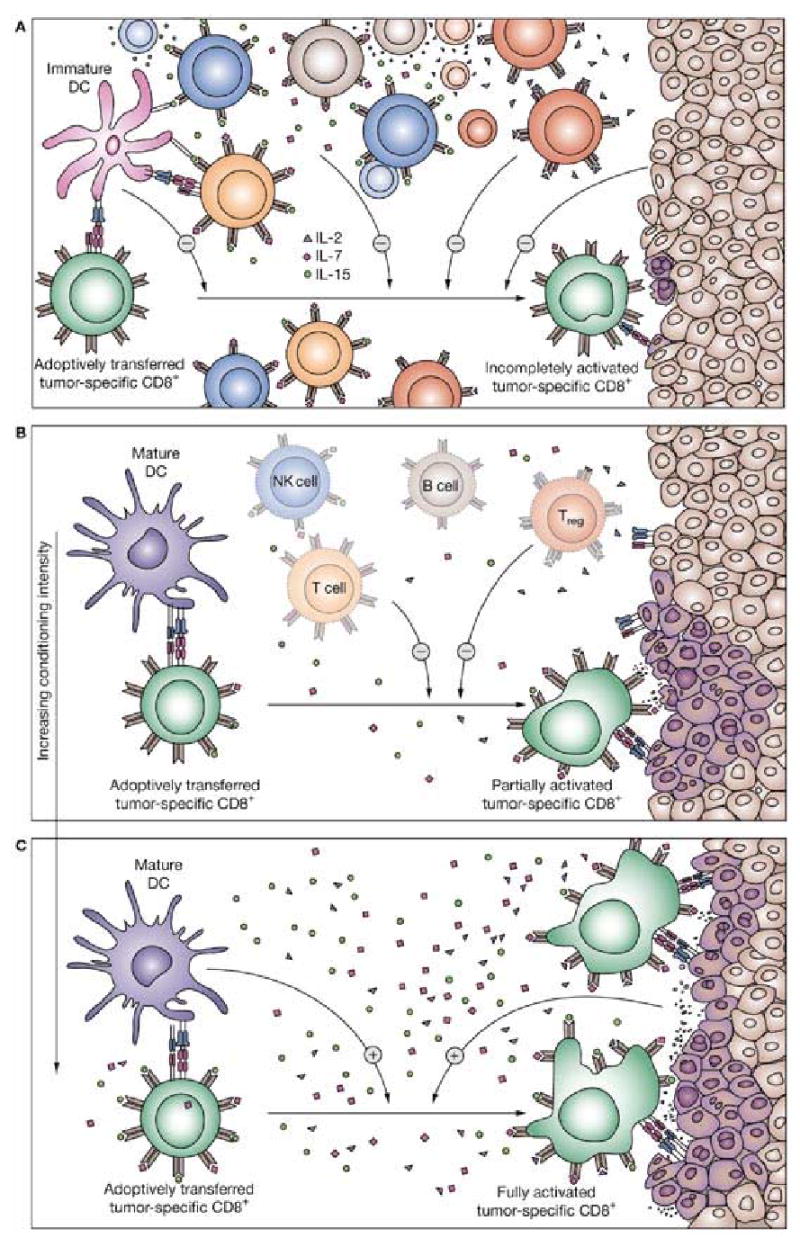
(A) In immunoreplete hosts, endogenous cells can inhibit adoptively transferred T cells by at least three mechanisms. (1) T cells, B cells and NK cells compete for homeostatic and activating cytokines such as IL-2, IL-7 and IL-15. (2) Treg and possibly NK cells and other immune cell populations (e.g. macrophages) display suppressive activity either by direct cell-to-cell contact or release of suppressive cytokines. (3) Immature DCs might fail to activate or might even anergize adoptively transferred T cells, limiting the antitumor response. (4) The tumor itself might act as a negative regulator of T-cell activation by actively producing regulatory molecules and by expressing only limited numbers of major histocompatibility complex–peptide complexes, rendering tumor recognition difficult.8 (B) Partial (nonmyeloablated) immunodepletion of the host with chemotherapy or total body irradiation before adoptive cell transfer reduces competing ‘cytokine sinks’ as well as regulatory elements. (C) Further increases of immunodepletion to levels that are myeloablative causes profound diminution of inhibitory elements and endogenous ‘cytokine sinks’. The proinflammatory environment activates immature DCs, although their numbers are further reduced. Moreover, conditioning regimen might also alter the tumor itself, promoting the expression of major histocompatibility complex–peptide complexes, increasing the pool of peptides available for presentation and the number of various costimulatory molecules.54 Thus, alteration of the host environment can lead to an enhanced activation of the transferred T cells, better recognition of the tumor, and ultimately more-efficient tumor destruction. Abbreviations: DC, dendritic cell; IL, interleukin; NK cells, natural killer cells; Treg, regulatory T cells.
Mere expansion of tumor-reactive T-cell numbers after a lymphodepleting preparative regimen does not seem to have had a critical role in our mouse model of cancer treatment.20 In fact, in the pmel-1 model the absolute number of tumor-specific T cells present in lymph nodes, spleen, blood and tumor recovered from treated mice was not different between irradiated and nonirradiated hosts. Significant differences were observed in the functional capacity of these T lymphocytes recovered from the irradiated host, and after antigen restimulation these cells produced greater amounts of the cytokines interferon-γ , IL-2, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor-α .22 Taken together, these observations suggest that the lymphopenic environment is associated with an overall decrease in T-cell activation threshold, and a bias towards autoimmunity.
Lymphodepletion with TBI also reduces CD4+CD25+FOXP3+ Treg, which probably contributes to the decrease in activation threshold of effector T cells.45 Treg have been shown to play a role in immune tolerance against tumors in both mouse systems and humans.46, 47, 48 Adoptive transfer of CD4+ T-helper cells selectively depleted of CD25+ populations indeed proved to increase antitumor response in the pmel-1 mouse model, while the infusion of CD4+CD25+ T cells (containing the FOXP3+ Treg population) had a profound negative impact on the efficacy of treatment.49
In addition to regulatory CD4+ lymphocytes, other cellular elements also influence the effectiveness of ACT. Surprisingly, in the work from Gattinoni et al.22 an already effective response in genetically lymphopenic RAG−/− mice (which do not have any T cells, including Treg) could still be enhanced by sublethal irradiation. Similar effects in the same hosts were achieved by the selective depletion of abundant natural killer cells using monoclonal antibodies. This finding suggests that elimination of potential competitor cells or ‘homeostatic sinks’ may influence the availability of cytokines critical for function of antitumor effectors.
TBI may not only lead to an increased lymphodepletion, but also affect T-cell trafficking, adhesion and costimulation. For example, in a murine model of graft versus host disease (GVHD), radiation caused upregulation of VCAM1, ICAM1, and B7-2, providing early costimulatory signals leading to priming of allogeneic T cells in the intestine.50 TBI could significantly alter the function of APCs. In a murine model of allogeneic transplant, TBI caused host dendritic cells (DCs) to undergo a rapid activation with upregulation of MHC class II and costimulatory molecules and secretion of IL-12, which is known to direct T-cell function towards cytotoxic type 1 phenotype and function.51 Even though host DC numbers were dramatically decreased following TBI, their initial transient activation was sufficient to cause allogeneic T-cell priming and an increase in the severity of GVHD. Similarly, radiation and chemotherapeutic agents are known to modulate immunogenicity of murine and human tumor cells.52 Local irradiation (10–20 Gy) has been shown to cause upregulation of MHC class I, FAS, CEA1, mucin 1 and other proteins in 21 out of 23 human carcinoma cell lines.53 In another recent report, ionizing radiation not only elevated initial expression of MHC class I, but also caused an increase in the pool of peptides available for presentation, further augmenting synthesis of new MHC class I complexes. The peptide pool was derived from initially damaged cellular structures as well as subsequent repair processes, which led to the generation of novel and unique sequences and an increase in tumor recognition.54 The importance of this local effect, however, remains unclear, as in our own model shielding the tumor during TBI did not affect the treatment outcome.22
An additional intriguing aspect of lymphodepletion is the possibility of combining this technique with vaccination strategies. Antigenic stimuli in the lymphopenic state may effectively shift the T-cell repertoire towards the antigen-specific cells.55 In a mouse model, the frequency of antimelanoma T cells was four times higher after vaccination of lymphodepleted animals with an actively expanding T-cell population than it was in lymphoreplete hosts, and these cells also had a higher cytotoxic potential both in vitro and in vivo.56 In another model, DCs pulsed with breast tumor lysate were more effective at producing a response when administered to lymphopenic mice with a homeostatically expanding T-cell population.57 The clinical relevance of this phenomenon was recently demonstrated in a randomized clinical trial in which high-dose chemotherapy and autologous stem-cell transplantation caused rapid and effective reconstitution of specific antimicrobial immunity after the early infusion of in vivo primed and in vitro expanded T cells.58
The above examples show how partial lymphoablation before ACT appears to improve treatment. Emerging experimental data indicate that increased-intensity conditioning or even complete (lethal) myeloablation followed by autologous (or syngeneic) hematopoetic stem-cell rescue might further enhance treatment efficacy.59 In our pmel-1 model, administration of a lethal dose of radiation overcomes the previous sine qua non requirement for a vaccine administration, an observation of significant clinical importance as currently we transfer TILs of unknown specificity for which clinical-grade vaccines are not available.60 The exact mechanism of this effect is still under investigation. More complete removal of Treg and depletion of ‘cytokine sinks’ and perhaps other regulatory elements play a role. Immune reconstitution after myeloablative conditioning and infusion of T-cell-depleted stem cells is relatively slow, particularly in the CD4+ compartment.61, 62, 63
It seems likely that full ablation can lead to an even more favorable ratio of effectors:inhibitory elements, thus removing an important obstacle to effector cell function.60 Another potential explanation for the success of full ablation is that high-dose TBI could simply lead to diffuse tissue injury and a generalized inflammatory reaction, which drives T-cell responses. Tissue injury generates ‘danger signals’,64 leading to the activation of DCs and other APCs. In the gastrointestinal tract TBI causes mucosal damage, leading to bacterial translocation and release of lipopolysaccharide, which might act via Toll-like receptors, further enhancing the function of APCs and promoting T-cell activation and expansion.65, 66, 67
Developing a safe lymphodepleting regimen for future trials
Nonmyeloablative lymphodepleting preconditioning with cyclophosphamide and fludarabine illustrated the importance of reducing the host lymphocyte pool prior to adoptive transfer of TILs.17 The above-described animal data may give a compelling reason to investigate a truly ‘lymphomyeloablative’ TBI-based regimen as a basis for further enhancement of ACT therapy. While it may be true that lymphodepletion can be of critical importance for the optimal function of transferred cells in the ACT setting, we need to consider the risks of this approach when applied to human subjects. Selection of an appropriate conditioning method appears to be crucial, as dose intensification increases regimen-associated toxicities.68, 69, 70 Drawing direct analogies with the regimens used for treating hematologic malignancies might not be optimal, as the goal of immune ablation prior to ACT for solid tumors is different. In fact, the aim of complete myeloablation for hematologic disease is not only the creation of appropriate conditions for engraftment, but also the direct elimination of the malignant population, whereas in ACT for solid tumors the objective is to induce lymphodepletion and perhaps an inflammatory state. Experience from auto-stem and allo-stem-cell transplantation in the setting of hematologic malignancy68, 71 or autoimmune disease72 clearly demonstrates that high-intensity conditioning can be associated with multiple toxicities, including prolonged neutropenia and the associated risk of infection, mucositis, graft failure, engraftment syndrome, and early and late effects of irradiation (e.g. diffuse alveolar hemorrhage, interstitial pneumonitis (IP), bronchiolitis obliterans, secondary myelodysplastic syndrome, secondary malignancy, cataracts, renal insufficiency, etc). TBI-related pulmonary complications are of significant concern and represent a major cause of mortality.73, 74 It is possible that pulmonary toxicity after ACT and autologous myeloablative transplantation should be lower than after allogeneic transplants, but variable data have been reported regarding the risk of IP in autologous settings.75, 76
The effects of TBI with cyclophosphamide and fludarabine in combination with IL-2 administration are unknown. Moreover, the safe and optimal dose of irradiation in the setting of stem-cell transplantation is still debatable. Toxicities frequently overlap, and combined effects of chemotherapy and radiotherapy are difficult to estimate; however, escalation of conditioning intensity can increase treatment-related morbidities. The addition of fractionated radiation (8.5–13.5 Gy) to a chemotherapy regimen used for autologous transplantation in patients with multiple myeloma resulted in treatment-related mortality (TRM) comparable to that with chemotherapy alone (2% versus 5%); however, the radiation group suffered 90% grade III–IV nonhematologic toxicity, compared with 65% in the nonirradiated group.77 Similarly, administration of TBI in some autoimmune diseases (e.g. scleroderma) has been associated with excessive toxicity, precluding further use of this method in this setting.78 The mean dose of radiation to the lung might have an independent predictive value with regard to pulmonary-toxicity-related mortality in the setting of both autologous and allogeneic stem-cell transplantation.76 Lung shielding appears to be relatively effective at reducing incidence of pulmonary toxicities, but its value is still in question.79 Shielding may prevent ablation of mediastinal immune organs, and may adversely impact the efficacy of treatment in hematologic malignancies, but this is likely to be less significant in the setting of solid tumors such as melanoma for which the primary goal of TBI is not direct tumor killing.
A retrospective analysis of the dose-response effect of 26 conditioning regimens on pulmonary toxicity from 20 reported studies that included 1,090 patients was recently reported.80 The authors identified a dose-response effect for both cyclophosphamide, and radiation dose and fractionation (Figure 2). They estimated that 120 mg/kg cyclophosphamide alone would be associated with a 3–4% risk of developing IP. A single radiation dose of 8.8 Gy with cyclophosphamide would be associated with 50% risk for IP, while a single dose of 5.1 Gy was estimated to carry a risk of less than 5%. A total irradiation dose of 12 Gy in 6 daily fractions would be associated with an 11% incidence of IP. Lung shielding of 50% could theoretically reduce the IP risk to 2.8%. Unfortunately, this model of logistic regression did not accommodate twice-daily fractionation, which is commonly used to further reduce pulmonary toxicity.
Figure 2. Dose–response functions using a mathematical model described by Sampath et al. based on data collected from clinical trials of bone marrow transplant80.
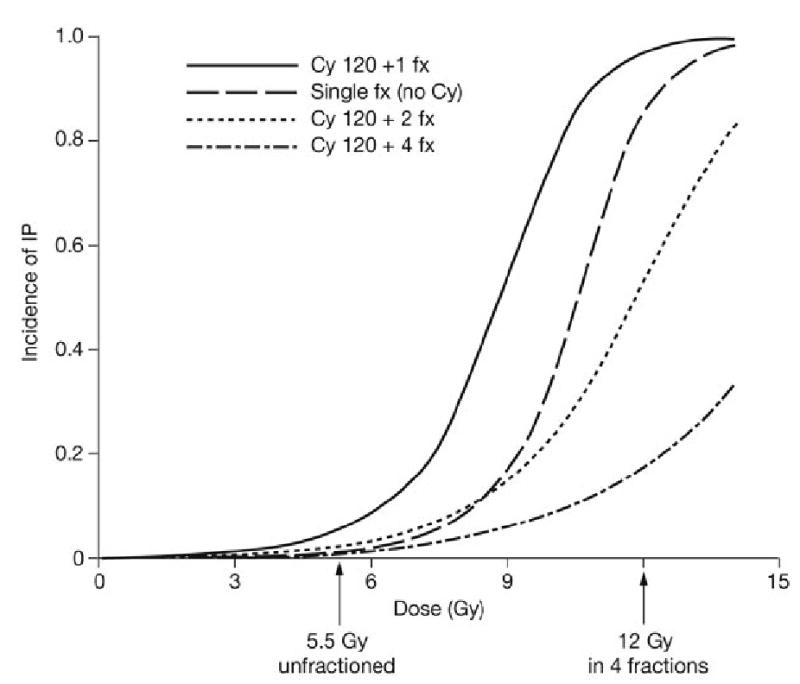
Dose–response functions are shown for 1, 2, and 4 fractions combined with 120 mg/kg Cy given over 2 days. A single-dose dose–response function without any Cy is also shown. The arrows highlight the doses of 5.5 Gy and 12 Gy. As shown in the graph, fractionation greatly decreases the incidence of IP at any given dose; in this mathematical model either a 5.5 Gy single dose or a 12 Gy fractionated dose appears to be associated with acceptable IP incidence. Lung shielding is expected to further decrease the risk of IP. Adapted from Elsevier Ltd © Sampath S et al. (2005) Int J Radiat Oncol Biol Phys 63: 876–884. Abbreviations: cy, cyclophosphamide; fx, fraction; IP, interstitial pneumonitis.
A protocol utilizing cyclophosphamide and fludarabine with TBI has been successfully used at the Hematology Branch of the National Heart Lung and Blood Institute (NHLBI) as a conditioning regimen for myeloablative T-cell-depleted allogeneic stem-cell transplantation for hematologic malignancy,79 and could serve as a potential platform for adoptive immunotherapy for solid tumors. Initially, this protocol consisted of 13.6 Gy of TBI and 120 mg/kg cyclophosphamide; in its latest iteration the protocol was modified to include 120 mg/kg cyclophosphamide, and 125 mg/m2 fludarabine with 12.0 Gy of TBI in 8 fractions with lung shielding (lung dose 9.0 Gy reduced later to 6.0 Gy). Since 1997, the TRM among 146 patients treated at the NHLBI with this regimen has been relatively low at 16% (n = 18). The majority of regimen-related fatalities occurred as a result of pulmonary transplant-related mortality (PTRM; 10.5%, n = 14). Causes of death were IP (n = 6), acute respiratory distress syndrome (n = 4), pneumonia (n = 4), cytomegalovirus (n = 2), respiratory syncytial virus (n = 1) and bacterial infection (n = 1). Of note, there was no PTRM reported in a series of 26 patients after 50% lung shielding was introduced into the regimen.79 A similar TBI dose was used in the setting of autologous hematopoetic stem-cell transplantation in 21 patients with advanced multiple sclerosis, for which the preparative regimen included 120 mg/kg cyclophosphamide and 12 Gy of TBI fractionated into 150 cGy twice daily with 50% lung shielding. No procedure-related mortality was observed; however, improvement in underlying disease was marginal.81
Alternatively, the reduced-intensity conditioning regimen can utilize a single 5.5 Gy dose of radiation delivered at a high rate (30 cGy/min).82 This regimen was designed with the intention of minimizing procedure-related toxicity in high-risk patients, while still allowing for efficient engraftment, as studies have demonstrated that an unfractionated single dose of radiation might be more immunosuppressive than a comparable dose administered at a slow rate or in fractions.83, 84 In a matched related allogeneic transplant setting, overall TRM was 7–19% at 2 years, depending on the risk group.82 In another high-risk cohort of 110 matched unrelated graft recipients, grade IV end-organ toxicities occurred in 5 patients;85, 86 overall TRM was 30% at 1 year, and 29 patients died of infection and 6 of GVHD.
The above data suggest that a protocol involving either a single 5.5 Gy dose of TBI or a fractionated dose of 10–12 Gy with 50% lung shielding and autologous CD34-selected stem-cell support might be relatively safe in terms of acute or subacute toxicity. It is possible, however, that other methods of irradiation (e.g. total lymphoid irradiation or its combination with TBI) could be equally immunosuppressive and safer. It is also not clear whether the coadministration of IL-2 immediately after such an irradiation is safe. It may be of concern that TBI and IL-2 affect endothelial permeability,78, 87 which could potentiate toxicities. Current experience from our institution demonstrates that lymphodepletion greatly improves tolerance to subsequent treatment with IL-2.88 Other, unexpected toxicities might occur at various organs compromised by radiation and neutropenia and could be further compounded by the cytokine and immune response. These adverse events may in particular affect patients previously exposed to other treatments for their underlying disease.
It will be crucial to perform appropriate initial screening of patients enrolled into such an aggressive protocol,89 but prediction of morbidity and mortality could be difficult.74 Exclusion criteria should be based on the criteria applied to allogeneic stem-cell transplantation candidates. Baseline diffusion capacity of the lung for carbon monoxide (DLCO <85% predicted) has been demonstrated to be the single most predictive factor for PTRM;79, 90 therefore, patients should undergo pulmonary function testing and be evaluated for history of tobacco use. In addition, extensive metastatic disease involving vital organs appears to pose additional risk of serious complications (e.g. bowel perforation, cardiac tamponade, or intracranial hemorrhage). Following administration of myeloablative chemoradiotherapy and transfer of T cells and stem cells, patients will require considerable supportive care and careful monitoring, as well as aggressive prevention and treatment of infectious and noninfectious complications, using protocols developed in the setting of hematopoetic stem-cell transplantation.
A platform for a new generation of adoptive immunotherapies
Administration of nonmyeloablative regimens before ACT has been shown in animal models and patients to substantially improve the tumor treatment efficacy of transferred tumor-reactive T cells.22 If an adoptive immunotherapy regimen employing ablative immunosuppressive preconditioning is translated successfully and safely from animal models into human patients, it could serve as a testing ground to explore a new generation of immune therapies by facilitating the use of alternative sources of effector cells, and would therefore help in addressing some key limitations that are associated with current TIL treatment. Autologous effector cells need to be generated from each patient’s tumor and expanded in vitro.21 This method greatly limits the number of patients that can be treated. Furthermore, the in vitro generation and expansion of TILs can be unsuccessful. Even if patients receive adequate infusion of cells, responses may fail to occur in a significant fraction of the treated patients. While many of the objective responses observed with current immunotherapies offer significant palliation to patients, partial responses are generally not durable or curative. One of the reasons for the failure could be that the autologous repertoire of T cells lack TCRs with high avidity towards self-antigens as a result of central tolerance.6 It is also clear that the cells used for ACT need to display certain characteristics to be effective in vivo, and the process of in vitro expansion might result in cell senescence. In our pmel-1 model the antitumor effectiveness of the transferred T cells correlated with the functional status; paradoxically, cells that were more activated and differentiated in vitro displayed less in vivo therapeutic efficiency then more-naive cells.91 A recent report in patients with metastatic melanoma showed that the persistence of the transferred cells is related to the length of the telomeres before transfer;92 ‘older’ cells are less likely to persist and to be therapeutically effective than ‘younger’ cells. In the light of these findings it might be difficult to produce optimal cells from everyone.
One could envision that the availability of ‘off-the-shelf’ third-party tumor-specific T cells could significantly simplify therapy, making ACT-based regimens much more effective and accessible.93 Allogeneic cells are, however, rapidly and efficiently rejected in immunocompetent hosts, limiting the survival of the transferred lymphocytes. In some studies the mean survival of white blood cells in patients receiving multiple transfusions was 2 days.94, 95 Administration of an aggressive lymphodepleting regimen before the transfer can prolong the persistence of these allogeneic lymphocytes. For example, haploidentical natural killer cells can expand to a detectable level in the peripheral blood only in patients treated with the most aggressive conditioning regimen (i.e. high-dose cyclophosphamide and fludarabine).96
Third-party effector lymphocytes could be generated by various methods; the simplest would be obtaining TILs from one patient (preferably a very good, complete responder) and using them for treatment of a different patient. Lymphocytes could be only partially matched in terms of HLA compatibility to ensure their function via recognition of restriction elements on APCs and tumor. This idea could evolve further into the use of allo-restricted tumor-specific cells generated from unrelated or related donors.97, 98 Mouse model data showed that such cells were relatively easy to obtain.99 Potentially, a library of T-cell clones against common cancer antigens could be developed and those cells rapidly tested against patients’ tumors and used for treatment (Figure 3).
Figure 3. Possible applications of adoptive cell transfer using a preconditioning lymphodepleting regimen consisting of chemoradiotherapy and the use of different types of donor T cells in conjunction with administration of exogenous cytokines.
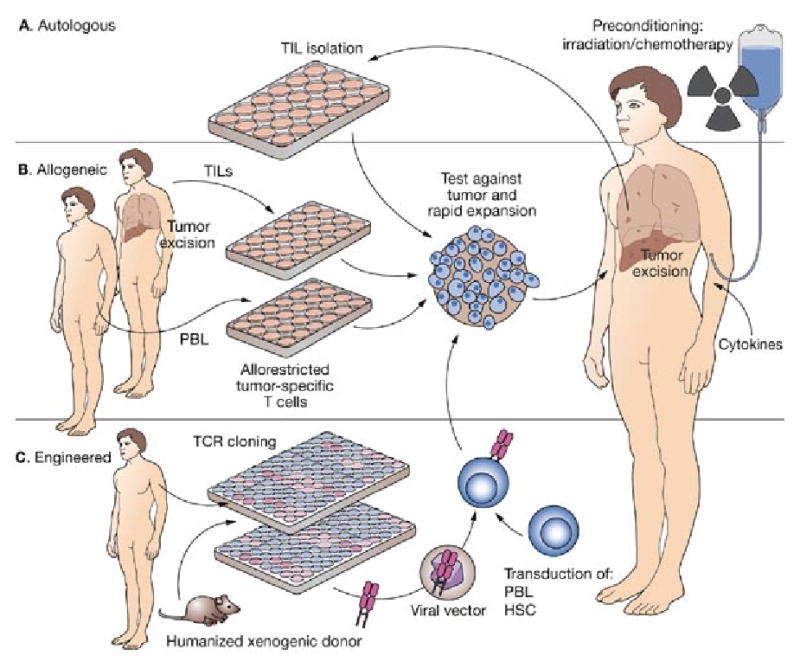
(A) The approach used by Dudley et al. in their recent trial.17 Autologous TILs were isolated from the tumor samples harvested from the patient, tested, expanded and reinfused. (B) The allogeneic cells transferred into the patient could be derived from TILs harvested from another patient (a good responder to autologous adoptive cell transfer) or generated in vitro from peripheral blood lymphocytes of an allogeneic healthy donor and selected for their ability to recognize tumor epitopes in the context of the recipient’s major histocompatibility complex. (C) Effector T cells generated by gene therapy methods using lentiviral or retroviral vectors. TCR sequences can be derived from either allogeneic or xenogenic sources.109, 112 In the case of xenogenic source, donor animals must express human restriction element (i.e. human leukocyte antigen allele presents in the patient) in order to be able to recognize human antigens on human tissues. Xenogenic donors have a T-cell repertoire not influenced by negative selection against human self-peptide; thus, it may be easier to generate cells with high-avidity TCR for human tumor antigens. In the case of an allogeneic donor a clone expressing a TCR reactive against the patient’s tumor can be generated either in vitro from a healthy donor (i.e. allogeneic allorestricted tumor-specific T cells) or from the TILs of another patient (see B). After isolation of tumor-reactive cells, the TCR is cloned and inserted into a viral vector and used to transduce either autologous or allogeneic PBL, peripheral or cord blood HSC,113 or even an immortalized T-cell line; the cells are then selected, matured and expanded as needed, and reinfused. In all these scenarios it is also possible to further manipulate cells before transfer by genetic means. This process may include insertion of sequences encoding for cytokines (e.g. interleukins 2 or 15), adhesion molecules, antiapoptotic or suicide genes, and so on.108 Abbreviations: HSC, hematopoietic stem cells; PBL, peripheral blood lymphocytes; TCR, T-cell receptor; TIL, tumor-infiltrating lymphocyte.
The feasibility of this approach has been demonstrated in eight solid organ recipients with refractory transplant-related EBV lymphoproliferative disease100, 101 who received third-party in vitro generated partially matched tumor-specific lymphocytes selected from an available library. There were four objective responses, including three complete remissions, and GVHD was not observed. Similarly, several clinical trials were performed in EBV-related diseases (nasopharyngeal carcinoma and Hodgkin’s disease) and in the bone marrow transplant setting using infusion of donor virus-specific T cells to prevent CMV infection.102, 103, 104
The potential risk of GVHD with allogeneic antitumor cells is likely to be less than in the case of nonspecific donor lymphocyte infusion, because ex vivo expanded tumor-specific T-cell preparations are likely to have T-cell clonotypes that are primarily directed against a somewhat limited set of ‘self’ tumor epitopes.105, 106 Furthermore, as an additional safety measure, autologous stem cells and lymphocytes could be used to rescue the host and eliminate transferred allogeneic cells should GVHD occur.
‘Off-the-shelf’ lymphocytes could also be entirely constructed in vitro by gene therapy methods.107, 108, 109 Effector cells could be transduced with lentiviral or retroviral constructs encoding for tumor-reactive TCRs. This technique would allow the use of engineered TCRs generated in vitro by phage display or in vivo in an allogeneic or even xenogeneic setting.110 For example, mice transgenic for human HLA molecules with a repertoire not influenced by negative selection in the thymus could contain high-avidity T cells with strong reactivity against human epitopes—and could be highly effective in therapeutic applications. Not only can melanoma-specific TCRs be obtained by this approach, but also a wide variety of tumor antigens can be targeted. For example, Sherman and colleagues have obtained TCR specific for human p53 by immunizing mice transgenic for the HLA-A*0201 molecule.111, 112 Since the transduced mature T cells already express endogenous α and β chains, the delivered α and β chain will mis-pair with these TCRs and thereby reduce their expression on the cell surface. One solution would be the in vitro maturation of T cells generated de novo from TCR-transduced peripheral or cord blood stem cells,113, 114 so that the expression of the endogenous TCRs would be reduced or prevented by allelic exclusion. The naive phenotype of very ‘young’ cells would facilitate their migration into the lymph nodes and tumor sites; these cells would have long telomeres and much greater proliferative capacity than the senescent memory cells used currently, and their persistence and therapeutic effectiveness would, therefore, most likely be enhanced.22, 92
Conclusions
Many questions regarding the future of immune therapy remain unanswered, but our understanding of immune regulation is growing continuously. Multiple redundant homeostatic mechanisms maintain a tight balance between responsiveness and tolerance, thereby protecting the host from uncontrolled immune responses against pathogens and potentially harmful autoimmunity. Changing the equilibrium of various immune system populations by inducing lymphopenia may result in a selective advantage being given to adoptively transferred T cells. Animal data demonstrating the benefit of lymphodepleting conditioning is dramatic and convincing.22 Recent clinical results using a nonmyeloablative preparative regimen confirmed the importance of lymphodepletion prior to ACT.17 New experimental data suggest that more-profound immune depletion might further augment the efficacy of ACT therapy.60
Obviously, increasing the intensity of the conditioning regimen goes against a recent trend established by the transplant community, which has focused on reducing treatment-related adverse effects by using a nonmyeloablative approach.72 One will never know a priori whether performing myeloablation would only add toxicity with significant morbidity and mortality without providing clinical benefit. Perhaps other, less toxic and more selective methods of lymphoid ablation not discussed in this Review could be explored and developed. Once we have a better understanding of all the mechanisms involved, we should be able to selectively influence arms of the immune response to achieve the desired clinical effect.115, 116
One might also argue that the use of optimal effector cells is crucial for the outcome of any immunotherapy, and that their lack is the main factor limiting the efficacy of therapy. The ideal T cell should display high avidity for its target antigen and be relatively ‘young’, to be able to proliferate and traffic to its target. It is quite possible that only improvements in all these aspects are required to lead us to significant progress. A strategy for more profound lymphodepletion could prove to be the new standard approach for ACT therapy and could enable the use of cellular effectors derived from novel sources, but it is obligatory to strike the right balance between effectiveness and tolerability, and safety and toxicity, of new therapies. Morgan et al. have recently reported that adoptive transfer of autologous T cells that were genetically modified with tumor-specific TCR resulted in significant durable persistence in 15 patients. Two recipients experienced an objective clinical response and in both of them high levels of transduced lymphocytes were observed 12 months after the infusion, thus strongly suggesting potential therapeutic value of this approach. 110
Key points
A 50% objective response rate was reported after a lymphodepleting conditioning regimen prior to adoptive cell transfer for patients with metastatic melanoma, and robust long-term persistence of the adoptively transferred cells was observed
Multiple homeostatic mechanisms regulate the number and functional status of T-cell lymphocytes in a normal host, and normopenic conditions might impair persistence and function of adoptively transferred antitumor effector lymphocytes
Lymphopenia creates selective advantage for some populations of T cells, eliminating regulatory T cells and competing cell populations (‘cytokine sinks’), and enhancing the availability of vital cytokines (e.g. interleukins 2, 7 and 15). T cells undergo homeostatic expansion under these conditions and increase their activation state
Animal data suggest that increased intensity immune ablation with stem-cell support further enhances the effectiveness of adoptive cell transfer; multiple safety issues have to be considered when designing an appropriate lymphoablative/immunosuppressive conditioning regimen
In the future, increased intensity immuno-depletion may serve as a platform for the use of allogeneic lymphocytes that are genetically engineered to have antitumor activity
Acknowledgments
This work was supported by the Clinical Research Center of the National Institutes of Health. The authors would like to thank Dr James C Yang for critical reading of the manscript and valuable suggestions. The authors declare that they have no financial conflicts of interest.
Footnotes
Competing interests
The authors declared no competing interests.
References
- 1.Rosenberg SA, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313 :1485–1492. doi: 10.1056/NEJM198512053132327. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271:907–913. [PubMed] [Google Scholar]
- 3.Dutcher JP, et al. A phase II study of interleukin-2 and lymphokine-activated killer cells in patients with metastatic malignant melanoma. J Clin Oncol. 1989;7:477–485. doi: 10.1200/JCO.1989.7.4.477. [DOI] [PubMed] [Google Scholar]
- 4.Legha SS, et al. Evaluation of interleukin-2 administered by continuous infusion in patients with metastatic melanoma. Cancer. 1996;77:89–96. doi: 10.1002/(SICI)1097-0142(19960101)77:1<89::AID-CNCR15>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 5.Rosenberg SA, et al. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10:909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Derbinski J, et al. Promiscuous gene expression in medullary thymic epithelial cells mirrors the peripheral self. Nat Immunol. 2001;2:1032–1039. doi: 10.1038/ni723. [DOI] [PubMed] [Google Scholar]
- 7.Zippelius A, et al. Thymic selection generates a large T cell pool recognizing a self-peptide in humans. J Exp Med. 2002;195:485–494. doi: 10.1084/jem.20011658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marincola FM, et al. Tumors as elusive targets of T-cell-based active immunotherapy. Trends Immunol. 2003;24:335–342. doi: 10.1016/s1471-4906(03)00116-9. [DOI] [PubMed] [Google Scholar]
- 9.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayyoub M, et al. Lack of tumor recognition by hTERT peptide 540–548-specific CD8(+) T cells from melanoma patients reveals inefficient antigen processing. Eur J Immunol. 2001;31:2642–2651. doi: 10.1002/1521-4141(200109)31:9<2642::aid-immu2642>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Rosenberg SA, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–6176. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 12.Rosenberg SA. Cancer immunotherapy comes of age. Nat Clin Pract Oncol. 2005;2:115. doi: 10.1038/ncponc0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gattinoni L, et al. Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol. 2006;6:383–393. doi: 10.1038/nri1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ross S, et al. Adoptive immunotherapy of hormone-refractory, stage D2 prostate cancer using ex vivo activated autologous T cells (autolymphocyte therapy): results from a pilot study. Biotechnol Ther. 1993;4:197–211. [PubMed] [Google Scholar]
- 15.Yee C, et al. Melanocyte destruction after antigen-specific immunotherapy of melanoma: direct evidence of t cell-mediated vitiligo. J Exp Med. 2000;192:1637–1644. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudley ME, et al. Adoptive transfer of cloned melanoma-reactive T lymphocytes for the treatment of patients with metastatic melanoma. J Immunother. 2001;24:363–373. doi: 10.1097/00002371-200107000-00012. [DOI] [PubMed] [Google Scholar]
- 17.Dudley ME, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298:850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes MS, et al. Transfer of a TCR gene derived from a patient with a marked antitumor response conveys highly active T-cell effector functions. Hum Gene Ther. 2005;16:457–472. doi: 10.1089/hum.2005.16.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klebanoff CA, et al. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol. 2005;26:111–117. doi: 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laport GG, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003;102:2004–2013. doi: 10.1182/blood-2003-01-0095. [DOI] [PubMed] [Google Scholar]
- 21.Dudley ME, et al. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–912. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Therasse P, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92:205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg SA, et al. Gene transfer into humans—immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323:570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 25.Dudley ME, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–2357. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mills CD, North RJ. Expression of passively transferred immunity against an established tumor depends on generation of cytolytic T cells in recipient: inhibition by suppressor T cells. J Exp Med. 1983;157:1448–1460. doi: 10.1084/jem.157.5.1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang LX, et al. Host lymphodepletion augments T cell adoptive immunotherapy through enhanced intratumoral proliferation of effector cells. Cancer Res. 2005;65:9547–9554. doi: 10.1158/0008-5472.CAN-05-1175. [DOI] [PubMed] [Google Scholar]
- 28.Overwijk WW, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–580. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Muranski P, et al. Mature CD4+ T cells perceive a positively selecting class II MHC/peptide complex in the periphery. J Immunol. 2000;164:3087–3094. doi: 10.4049/jimmunol.164.6.3087. [DOI] [PubMed] [Google Scholar]
- 30.Kirberg J, et al. Peripheral T cell survival requires continual ligation of the T cell receptor to major histocompatibility complex-encoded molecules. J Exp Med. 1997;186:1269–1275. doi: 10.1084/jem.186.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kondrack RM, et al. Interleukin 7 regulates the survival and generation of memory CD4 cells. J Exp Med. 2003;198:1797–1806. doi: 10.1084/jem.20030735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ku CC, et al. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science. 2000;288:675–678. doi: 10.1126/science.288.5466.675. [DOI] [PubMed] [Google Scholar]
- 33.Schluns KS, et al. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. doi: 10.1038/80868. [DOI] [PubMed] [Google Scholar]
- 34.Goldrath AW, et al. Cytokine requirements for acute and Basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med. 2002;195:1515–1522. doi: 10.1084/jem.20020033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng R, et al. Synergy of IL-21 and IL-15 in regulating CD8+ T cell expansion and function. J Exp Med. 2005;201:139–148. doi: 10.1084/jem.20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakaguchi S, et al. Organ-specific autoimmune diseases induced in mice by elimination of T cell subset. I: evidence for the active participation of T cells in natural self-tolerance; deficit of a T cell subset as a possible cause of autoimmune disease. J Exp Med. 1985;161:72–87. doi: 10.1084/jem.161.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goldrath AW, et al. Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J Exp Med. 2000;192:557–564. doi: 10.1084/jem.192.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Prlic M, et al. Homeostatic expansion occurs independently of costimulatory signals. J Immunol. 2001;167:5664–5668. doi: 10.4049/jimmunol.167.10.5664. [DOI] [PubMed] [Google Scholar]
- 39.Klebanoff CA, et al. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hinrichs CS, et al. Programming CD8+ T cells for effective immunotherapy. Curr Opin Immunol. 2006;18:363–370. doi: 10.1016/j.coi.2006.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Powrie F, et al. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 42.King C, et al. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 43.Neujahr D, Turka LA. Lymphocyte depletion as a barrier to immunological tolerance. Contrib Nephrol. 2005;146:65–72. doi: 10.1159/000082066. [DOI] [PubMed] [Google Scholar]
- 44.Dummer W, et al. T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest. 2002;110:185–192. doi: 10.1172/JCI15175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Antony PA, et al. Interleukin-2-dependent mechanisms of tolerance and immunity in vivo. J Immunol. 2006;176:5255–5266. doi: 10.4049/jimmunol.176.9.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Turk MJ, et al. Concomitant tumor immunity to a poorly immunogenic melanoma is prevented by regulatory T cells. J Exp Med. 2004;200:771–782. doi: 10.1084/jem.20041130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Curiel TJ, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- 48.Antony PA, Restifo NP. CD4+CD25+ T regulatory cells, immunotherapy of cancer, and interleukin-2. J Immunother. 2005;28:120–128. doi: 10.1097/01.cji.0000155049.26787.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Antony PA, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–2601. doi: 10.4049/jimmunol.174.5.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eyrich M, et al. Sequential expression of adhesion and costimulatory molecules in graft-versus-host disease target organs after murine bone marrow transplantation across minor histocompatibility antigen barriers. Biol Blood Marrow Transplant. 2005;11:371–382. doi: 10.1016/j.bbmt.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, et al. Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J Clin Invest. 2002;109:1335–1344. doi: 10.1172/JCI14989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gelbard A, et al. Combination chemotherapy and radiation of human squamous cell carcinoma of the head and neck augments CTL-mediated lysis. Clin Cancer Res. 2006;12:1897–1905. doi: 10.1158/1078-0432.CCR-05-1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garnett CT, et al. Sublethal irradiation of human tumor cells modulates phenotype resulting in enhanced killing by cytotoxic T lymphocytes. Cancer Res. 2004;64:7985–7994. doi: 10.1158/0008-5472.CAN-04-1525. [DOI] [PubMed] [Google Scholar]
- 54.Reits EA, et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J Exp Med. 2006;203:1259–1271. doi: 10.1084/jem.20052494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ma J, et al. Anti-tumor T cell response and protective immunity in mice that received sublethal irradiation and immune reconstitution. Eur J Immunol. 2003;33:2123–2132. doi: 10.1002/eji.200324034. [DOI] [PubMed] [Google Scholar]
- 56.Hu HM, et al. Development of antitumor immune responses in reconstituted lymphopenic hosts. Cancer Res. 2002;62:3914–3919. [PubMed] [Google Scholar]
- 57.Asavaroengchai W, et al. Tumor lysate-pulsed dendritic cells can elicit an effective antitumor immune response during early lymphoid recovery. Proc Natl Acad Sci USA. 2002;99:931–936. doi: 10.1073/pnas.022634999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rapoport AP, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11:1230–1237. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 59.Wrzesinski C, Restifo NP. Less is more: lymphodepletion followed by hematopoietic stem cell transplant augments adoptive T-cell-based anti-tumor immunotherapy. Curr Opin Immunol. 2005;17:195–201. doi: 10.1016/j.coi.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wrzesinski C, et al. Hematopoietic stem cells promote the expansion and function of adoptively transferred anti-tumor CD8+ T cells. J Clin Invest. doi: 10.1172/JCI30414. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dumont-Girard F, et al. Reconstitution of the T-cell compartment after bone marrow transplantation: restoration of the repertoire by thymic emigrants. Blood. 1998;92:4464–4471. [PubMed] [Google Scholar]
- 62.Roux E, et al. Recovery of immune reactivity after T-cell-depleted bone marrow transplantation depends on thymic activity. Blood. 2000;96:2299–2303. [PubMed] [Google Scholar]
- 63.Heitger A, et al. Essential role of the thymus to reconstitute naive (CD45RA+) T-helper cells after human allogeneic bone marrow transplantation. Blood. 1997;90:850–857. [PubMed] [Google Scholar]
- 64.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 65.Matsumoto T, et al. Significance of bacterial flora in abdominal irradiation-induced inhibition of lung metastases. Cancer Res. 1988;48:3031–3034. [PubMed] [Google Scholar]
- 66.Kieper WC, et al. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. J Immunol. 2005;174:3158–3163. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]
- 67.Hill GR, et al. Total body irradiation and acute graft-versus-host disease: the role of gastrointestinal damage and inflammatory cytokines. Blood. 1997;90:3204–3213. [PubMed] [Google Scholar]
- 68.Sorror ML, et al. Comparing morbidity and mortality of HLA-matched unrelated donor hematopoietic cell transplantation after nonmyeloablative and myeloablative conditioning: influence of pretransplantation comorbidities. Blood. 2004;104:961–968. doi: 10.1182/blood-2004-02-0545. [DOI] [PubMed] [Google Scholar]
- 69.Diaconescu R, et al. Morbidity and mortality with nonmyeloablative compared with myeloablative conditioning before hematopoietic cell transplantation from HLA-matched related donors. Blood. 2004;104:1550–1558. doi: 10.1182/blood-2004-03-0804. [DOI] [PubMed] [Google Scholar]
- 70.Abraham R, et al. Intensification of the stem cell transplant induction regimen results in increased treatment-related mortality without improved outcome in multiple myeloma. Bone Marrow Transplant. 1999;24:1291–1297. doi: 10.1038/sj.bmt.1702060. [DOI] [PubMed] [Google Scholar]
- 71.Scott BL, et al. Myeloablative vs nonmyeloablative allogeneic transplantation for patients with myelodysplastic syndrome or acute myelogenous leukemia with multilineage dysplasia: a retrospective analysis. Leukemia. 2005;20:128–135. doi: 10.1038/sj.leu.2404010. [DOI] [PubMed] [Google Scholar]
- 72.Griffith LM, et al. Feasibility of allogeneic hematopoietic stem cell transplantation for autoimmune disease: position statement from a National Institute of Allergy and Infectious Diseases and National Cancer Institute-Sponsored International Workshop, Bethesda, MD, March 12 and 13, 2005. Biol Blood Marrow Transplant. 2005;11:862–870. doi: 10.1016/j.bbmt.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 73.Moreau P, et al. Comparison of 200 mg/m(2) melphalan and 8 Gy total body irradiation plus 140 mg/m(2) melphalan as conditioning regimens for peripheral blood stem cell transplantation in patients with newly diagnosed multiple myeloma: final analysis of the Intergroupe Francophone du Myelome 9502 randomized trial. Blood. 2002;99:731–735. doi: 10.1182/blood.v99.3.731. [DOI] [PubMed] [Google Scholar]
- 74.Chen CI, et al. Radiation-associated pneumonitis following autologous stem cell transplantation: predictive factors, disease characteristics and treatment outcomes. Bone Marrow Transplant. 2001;27:177–182. doi: 10.1038/sj.bmt.1702771. [DOI] [PubMed] [Google Scholar]
- 75.Kantrow SP, et al. Idiopathic pneumonia syndrome: changing spectrum of lung injury after marrow transplantation. Transplantation. 1997;63:1079–1086. doi: 10.1097/00007890-199704270-00006. [DOI] [PubMed] [Google Scholar]
- 76.Della Volpe A, et al. Lethal pulmonary complications significantly correlate with individually assessed mean lung dose in patients with hematologic malignancies treated with total body irradiation. Int J Radiat Oncol Biol Phys. 2002;52:483–488. doi: 10.1016/s0360-3016(01)02589-5. [DOI] [PubMed] [Google Scholar]
- 77.Barlogie B, et al. Total therapy with tandem transplants for newly diagnosed multiple myeloma. Blood. 1999;93:55–65. [PubMed] [Google Scholar]
- 78.Burt RK, et al. The rationale behind autologous autoimmune hematopoietic stem cell transplant conditioning regimens: concerns over the use of total-body irradiation in systemic sclerosis. Bone Marrow Transplant. 2004;34:745–751. doi: 10.1038/sj.bmt.1704671. [DOI] [PubMed] [Google Scholar]
- 79.Savani BN, et al. Prediction and prevention of transplant-related mortality from pulmonary causes after total body irradiation and allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2005;11:223–230. doi: 10.1016/j.bbmt.2004.12.328. [DOI] [PubMed] [Google Scholar]
- 80.Sampath S, et al. Dose response and factors related to interstitial pneumonitis after bone marrow transplant. Int J Radiat Oncol Biol Phys. 2005;63:876–884. doi: 10.1016/j.ijrobp.2005.02.032. [DOI] [PubMed] [Google Scholar]
- 81.Burt RK, et al. Hematopoietic stem cell transplantation for progressive multiple sclerosis: failure of a total body irradiation-based conditioning regimen to prevent disease progression in patients with high disability scores. Blood. 2003;102:2373–2378. doi: 10.1182/blood-2003-03-0877. [DOI] [PubMed] [Google Scholar]
- 82.Blum W, et al. Low-dose (550 cGy), single-exposure total body irradiation and cyclophosphamide: consistent, durable engraftment of related-donor peripheral blood stem cells with low treatment-related mortality and fatal organ toxicity. Biol Blood Marrow Transplant. 2002;8:608–618. doi: 10.1053/bbmt.2002.v8.abbmt080608. [DOI] [PubMed] [Google Scholar]
- 83.Storb R, et al. Fractionated versus single-dose total body irradiation at low and high dose rates to condition canine littermates for DLA-identical marrow grafts. Blood. 1994;83:3384–3389. [PubMed] [Google Scholar]
- 84.Storb R, et al. Comparison of fractionated to single-dose total body irradiation in conditioning canine littermates for DLA-identical marrow grafts. Blood. 1989;74:1139–1143. [PubMed] [Google Scholar]
- 85.Girgis M, et al. Chimerism and clinical outcomes of 110 recipients of unrelated donor bone marrow transplants who underwent conditioning with low-dose, single-exposure total body irradiation and cyclophosphamide. Blood. 2005;105:3035–3041. doi: 10.1182/blood-2003-07-2346. [DOI] [PubMed] [Google Scholar]
- 86.Hallemeier C, et al. Outcomes of adults with acute myelogenous leukemia in remission given 550 cGy of single-exposure total body irradiation, cyclophosphamide, and unrelated donor bone marrow transplants. Biol Blood Marrow Transplant. 2004;10:310–319. doi: 10.1016/j.bbmt.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 87.Vatistas S, Hornsey S. Radiation-induced protein loss into the gastrointestinal tract. Br J Radiol. 1966;39:547–550. doi: 10.1259/0007-1285-39-463-547. [DOI] [PubMed] [Google Scholar]
- 88.Dudley ME, et al. A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother. 2002;25:243–251. doi: 10.1097/01.CJI.0000016820.36510.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sorror ML, et al. Hematopoietic cell transplantation (HCT)-specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. 2005;106:2912–2919. doi: 10.1182/blood-2005-05-2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Crawford SW, Fisher L. Predictive value of pulmonary function tests before marrow transplantation. Chest. 1992;101:1257–1264. doi: 10.1378/chest.101.5.1257. [DOI] [PubMed] [Google Scholar]
- 91.Gattinoni L, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–1626. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhou J, et al. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175:7046–7052. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Duval L, et al. Adoptive transfer of allogeneic cytotoxic T lymphocytes equipped with a HLA-A2 restricted MART-1 T-cell receptor: a phase I trial in metastatic melanoma. Clin Cancer Res. 2006;12:1229–1236. doi: 10.1158/1078-0432.CCR-05-1485. [DOI] [PubMed] [Google Scholar]
- 94.Adams PT, et al. Detection of circulating donor white blood cells in patients receiving multiple transfusions. Blood. 1992;80:551–555. [PubMed] [Google Scholar]
- 95.Lee TH, et al. Transient increase in circulating donor leukocytes after allogeneic transfusions in immunocompetent recipients compatible with donor cell proliferation. Blood. 1995;85:1207–1214. [PubMed] [Google Scholar]
- 96.Miller JS, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 97.Dutoit V, et al. Functional analysis of HLA-A*0201/Melan-A peptide multimer+ CD8+ T cells isolated from an HLA-A*0201- donor: exploring tumor antigen allorestricted recognition. Cancer Immun. 2002;2:7. [PubMed] [Google Scholar]
- 98.Pittet MJ, et al. Ex vivo characterization of allo-MHC-restricted T cells specific for a single MHC-peptide complex. J Immunol. 2006;176:2330–2336. doi: 10.4049/jimmunol.176.4.2330. [DOI] [PubMed] [Google Scholar]
- 99.Savage P, et al. Use of B cell-bound HLA-A2 class I monomers to generate high-avidity, allo-restricted CTLs against the leukemia-associated protein Wilms tumor antigen. Blood. 2004;103:4613–4615. doi: 10.1182/blood-2003-11-3903. [DOI] [PubMed] [Google Scholar]
- 100.Haque T, et al. Reconstitution of EBV-specific T cell immunity in solid organ transplant recipients. J Immunol. 1998;160:6204–6209. [PubMed] [Google Scholar]
- 101.Haque T, et al. Treatment of Epstein-Barr-virus-positive post-transplantation lymphoproliferative disease with partly HLA-matched allogeneic cytotoxic T cells. Lancet. 2002;360:436–442. doi: 10.1016/S0140-6736(02)09672-1. [DOI] [PubMed] [Google Scholar]
- 102.Comoli P, et al. Adoptive transfer of allogeneic Epstein-Barr virus (EBV)-specific cytotoxic T cells with in vitro antitumor activity boosts LMP2-specific immune response in a patient with EBV-related nasopharyngeal carcinoma. Ann Oncol. 2004;15:113–117. doi: 10.1093/annonc/mdh027. [DOI] [PubMed] [Google Scholar]
- 103.Walter EA, et al. Reconstitution of cellular immunity against cytomegalovirus in recipients of allogeneic bone marrow by transfer of T-cell clones from the donor. N Engl J Med. 1995;333:1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 104.Rooney CM, et al. Treatment of relapsed Hodgkin’s disease using EBV-specific cytotoxic T cells. Ann Oncol. 1998;9 (Suppl 5):S129–S132. doi: 10.1093/annonc/9.suppl_5.s129. [DOI] [PubMed] [Google Scholar]
- 105.Whitelegg A, Barber LD. The structural basis of T-cell allorecognition. Tissue Antigens. 2004;63:101–108. doi: 10.1111/j.1399-0039.2004.00188.x. [DOI] [PubMed] [Google Scholar]
- 106.Whitelegg AM, et al. Investigation of peptide involvement in T cell allorecognition using recombinant HLA class I multimers. J Immunol. 2005;175:1706–1714. doi: 10.4049/jimmunol.175.3.1706. [DOI] [PubMed] [Google Scholar]
- 107.Stanislawski T, et al. Circumventing tolerance to a human MDM2-derived tumor antigen by TCR gene transfer. Nat Immunol. 2001;2:962–970. doi: 10.1038/ni1001-962. [DOI] [PubMed] [Google Scholar]
- 108.Kershaw MH, et al. Supernatural T cells: genetic modification of T cells for cancer therapy. Nat Rev Immunol. 2005;5:928–940. doi: 10.1038/nri1729. [DOI] [PubMed] [Google Scholar]
- 109.Morgan RA, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Voss RH, et al. Designing TCR for cancer immunotherapy. Methods Mol Med. 2005;109:229–256. doi: 10.1385/1-59259-862-5:229. [DOI] [PubMed] [Google Scholar]
- 111.Kuball J, et al. Cooperation of human tumor-reactive CD4+ and CD8+ T cells after redirection of their specificity by a high-affinity p53A2.1-specific TCR. Immunity. 2005;22:117–129. doi: 10.1016/j.immuni.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 112.Cohen CJ, et al. Recognition of fresh human tumor by human peripheral blood lymphocytes transduced with a bicistronic retroviral vector encoding a murine anti-p53 TCR. J Immunol. 2005;175:5799–5808. doi: 10.4049/jimmunol.175.9.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schmitt TM, et al. Induction of T cell development and establishment of T cell competence from embryonic stem cells differentiated in vitro. Nat Immunol. 2004;5:410–417. doi: 10.1038/ni1055. [DOI] [PubMed] [Google Scholar]
- 114.Clark RA, et al. Human skin cells support thymus-independent T cell development. J Clin Invest. 2005;115:3239–3249. doi: 10.1172/JCI24731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Boyman O, et al. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006;311:1924–1927. doi: 10.1126/science.1122927. [DOI] [PubMed] [Google Scholar]
- 116.Gattinoni L, et al. CTLA-4 dysregulation of self/tumor-reactive CD8+ T cell function is CD4+ T cell-dependent. Blood. 2006 doi: 10.1182/blood-2006-07-034066. [doi: doi: 10.1182/blood-2006-07-034066] [DOI] [PMC free article] [PubMed] [Google Scholar]