

Abstract
The N1325S mutation in the cardiac sodium channel gene SCN5A causes the type-3 long QT syndrome but the arrhythmogenic trigger associated with N1325S has not been characterized. In this study, we investigated the triggers for cardiac events in the expanded N1325S family. Among 11 symptomatic patients with document triggers, six died suddenly during sleep or while sitting (bradycardia-induced trigger) and three died suddenly and two developed syncope due to stress and excitement (non-bradycardia-induced). Patch-clamping studies revealed that the late sodium current (INa,L) generated by mutation N1325S in ventricular myocytes from TG-NS/LQT3 mice, was reduced with increased pacing, which explains bradycardia-induced mortalities in the family. The non-bradycardic triggers are related to the finding that APD became prolonged and unstable at increasing rates, often with alternating repolarization phases which was corrected with verapamil. This implies that Ca2+ influx and intracellular Ca2+ ([Ca2+]i) ions are involved and that [Ca2+]i inhomogeneity may be the underlying mechanisms behind non-bradycardia LQT3 arrhythmogenesis associated with mutation N1325S.
Keywords: type 3 long QT syndrome (LQTS), ventricular arrhythmias, sudden death, sodium channel gene SCN5A, late persistent sodium current, APD, intracellular Ca2+
Introduction
The gain-of-function mutations in the cardiac sodium channel gene SCN5A cause type-3 long QT syndrome (LQT3), a clinical syndrome that is characterized by electrocardiographic prolonged QT intervals, high incidence of ventricular tachycardia (VT), ventricular fibrillation (VF), syncope and sudden death (LQTS) [1, 2]. To date, more than 80 LQTS mutations have been identified in SCN5A, but few have been characterized in cardiomyocytes. We expressed the N1325S mutation in the mouse heart by the cardiac-specific mouse α-myosin heavy chain promoter in a transgenic over-expression model [3]. Nuyens et al. [4] expressed ΔKPQ mutation in the heart by creation of a knockin mouse model. One common similarity shared by these two models is the development of the LQT3 phenotype. However, the incidences of arrhythmias in mice that express the N1325S mutation occur during non-bradycardia conditions (i.e., during elevated heart rates or increased pacing stimulation) [3]. In contrast, excessive bradycardia must be present in ΔKPQ mice to effectively predispose the mice to higher incidence of cardiac arrhythmias [4]. It is intriguing to speculate that such differences may reflect the variability that is present in LQT3 patients with different mutations. It is well known that in LQT3 patients there is an association between the incidence of cardiac events and heart rates, such that cardiac arrhythmias are often initiated by either heart rate deceleration or acceleration [5, 6]. However, no detailed genotype-phenotype correlation studies on different event-triggers were reported for different SCN5A mutations. In this study, we report the triggers for cardiac arrhythmias and sudden death associated with mutation N1325S. Furthermore, electrophysiological studies were carried out in TG-NS/LQT3 cardiomyocytes to investigate the mechanisms for the dual effects of heart rate on LQT3 arrhythmogenesis.
Materials and Methods
All experiments were conducted in accordance with the guidelines of the Cleveland Clinic Foundation Institutional Review Board on Animal Subjects and conformed to NIH guidelines.
Isolation of ventricular myocytes
Details for the isolation procedure have been previously reported [3], [7]. In brief, hearts from age-matched (6–8 months) adult non-transgenic (NTG) and the transgenic LQT3 (TG-NS/LQT3) mice were dispersed enzymatically and isolated by the Langendorff perfusion. Hearts were perfused a Ca2+-free with 0.03 mM EGTA, 0.15 mg/ml bovine serum albumin (BSA, Sigma), 20 U protease (Type XXIV, Sigma), and 0.06 mg/ml collagenase (Type II, 209 U/mg, Worthington) for approximately 28 mins followed by a step-wise increase in Ca2+ concentration (to 1.2 mM). Ventricular tissue was removed, placed in incubation buffer and gently triturated. Final cells were collected by gravity sedimentation and re-suspended in recording bath solution.
Whole-cell voltage and current recordings
Single, quiescent, and rod-shaped myocytes were chosen and experiments were conducted using a MultiClamp 700A amplifier interfaced to a Pentium computer equipped with Digidata 1322A and pClamp 9 software (Axon Instruments). Pipettes were fabricated from borosilicate glass capillaries (FHC) and fire-polished with 1.8–2.3 MΩ resistance. Signals were low-passed filtered at 1 kHz and digitized at 10 kHz and current recordings were not corrected for leak. Cells were held at −80 mV and the specific clamp protocols are indicated with the data. For current-clamp recordings, cells were depolarized with a current step of 1–1.5 nA and 25–40 ms duration at the specified cycle lengths (CLs). Cell capacitance was measured by calculating the area under the capacitive transient elicited by a −10 mV pulse. All recordings were made at room temperature. Data were fit to model equations using non-linear regression with pClamp 9.0 or Sigma Plot ver. 8.0. and the specific formulas are indicated in the results.
Solutions
Perfusion Solution I buffer contained the following reagents (in mM): 118, NaCl; 4.8, KCl; 1.8, CaCl2; 2.5, MgCl2; 1.2, KH2PO4; 11, glucose; 13.8 NaHCO3; 4.9, pyruvic acid; pH 7.2–7.4 with 95%O2:5%CO2; 37°C. For adequate voltage control of INa currents, a low external-(Na+) bath solution was used consisting of (in mM): 40, NaCl; 110, CsCl, 1.2, CaCl2; 2, CdCl; 2, MgCl2; 10, HEPES; 10, glucose; pH 7.3 with 1 M CsOH. To reduce contaminating K-currents, 3 mM 4-aminopyridine was freshly prepared and added to the external bath prior to the start of each experiment. The internal pipette solution was composed of (in mM): 130, CsCl; 10, NaCl; 5, EGTA; 10 HEPES; 10, glucose; pH 7.2 with 1 M CsOH. For current-clamp recordings, external buffer was (in mM): NaCl, 140; KCl, 4; CaCl2, 1.2; MgCl2, 2; HEPES, 10; glucose, 10; pH 7.2 with 5N NaOH, whereas internal pipette solution was of the following composition (in mM): KCl, 135; MgCl2, 1; EGTA 10; HEPES, 10; glucose, 10; pH 7.2 with 1N KOH.
Statistical Analysis
Data are expressed as mean ± SEM. (or as SD where indicated). Statistical significance was determined using the Student t test for comparison of two means and differences were considered statistically significant with a probability value of P < 0.05.
Results and Discussion
Multiple triggers precipitate cardiac events in a large N1325S LQTS family
In the human population, two distinct LQT3 sub-groups exist and are classified by arrhythmia susceptibility according to the patient’s physiological heart rate, i.e., bradycardic or non-bradycardic, which is often a clinical predilection for their arrhythmogenic episodes. Interestingly, LQTS patients in a single family with the SCN5A N3125S mutation responded to both bradycardic and non-bradycardic triggers.
The N1325S mutation was originally identified by Wang et al. [8] in a very small family with LQTS. In this study, we have now expanded the family to 96 members suitable for genotype-phenotype correlation studies. The average QTc for mutation carriers was 455.8 ± 9.7 ms, PR interval was 138.6 ± 9.2 ms, and QRS duration was 86.2 ± 4.3 ms. The ventricular rate was 85.8 ± 4.0 bpm for mutation carriers, and 70 bpm for the normal family member in the family. Twenty-one family members had sudden death, and two developed syncope episodes. Bradycardic triggers were identified in 26% of patients with cardiac events, 5 died suddenly during sleep, and one died suddenly due to sitting (Fig. 1). Non-bradycardic triggers were recorded in 22% of symptomatic patients, in which 3 died from stress-related excitement, and 2 developed syncope episodes from stress. Fifteen family members died suddenly, but no known triggers were documented (52% of cases, Fig. 1). One of the male mutation carriers developed a dilated left atria and left ventricle, atrial fibrillation, mitral valve prolapse, concentric left ventricular hypertrophy, and marked first degree AV block on ECG. Mitral valve prolapse was identified in two other female mutation carriers.
Fig. 1.
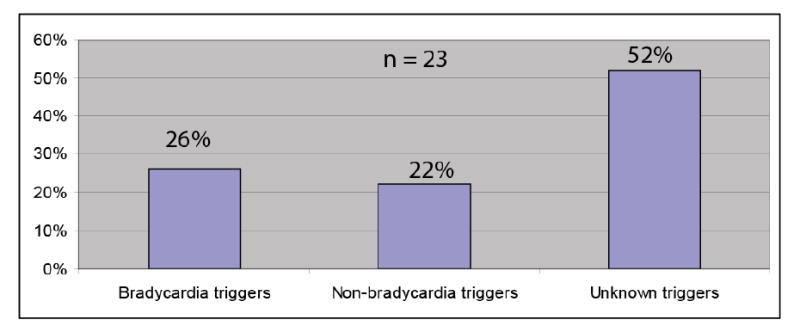
Triggers for cardiovascular events in a LQT3 family with SCN5A mutation N1325S. A total of 23 family members developed LQTS-associated symptoms, 21 sudden deaths and 2 cases of syncope.
Reduction of late INa currents generated by mutation N1325S with increasing pacing rate
To determine the mechanism by which the N1325S mutation causes bradycardia-induced VT and sudden death, we performed electrophysiological studies of INa,L with different stimulation rates. Fig. 2A shows 50 consecutive INa currents elicited by 50 ms square steps from −80 to −20 mV at 5 Hz from NTG and TG-NS/LQT3 single ventricular myocytes. Mutations in the regions involved in the inactivation process allow repeated channel reopening during prolonged depolarization, evoking a late and sustained INa current [9]. For the NTG cell, a large fraction of the current was inactivated after 5 ms, whereas for the TG-NS/LQT3 cell, a larger and sustained component decayed much slower over the same time course. The insets represent magnified portions of the decaying sodium current between 5–20 ms and reveal a non-inactivating component in the TG-NS/LQT3 but not in the NTG cell. This non-inactivating component or INa,L was decreased with each successive stimulation step. Since INa,L generated by LQTS mutations has been proposed as the main determinant for LQT3, its increase with decreasing rates is the likely mechanism for bradycardia-induced cardiac events in LQT3, as proposed by Nagatomo et al. [10]. In both cell groups, the declining phase of this component was fitted with a biexponential function (Fig. 2, legend) and the slow time component, τ2, was plotted as a function of pulse numbers (Fig. 2B). Peak INa was measured as the peak current amplitude remaining relative to the 1st pulse (Fig. 2C). As a function of rate, both INa,L and peak INa were decreased with increasing stimulation frequency, but the effect was more prevalent in TG-NS/LQT3 cells. The further decrease in peak INa in transgenic cells may suggest that decreases in membrane excitability at high stimulation rates may also contribute to arrhythmogenesis.
Fig. 2.
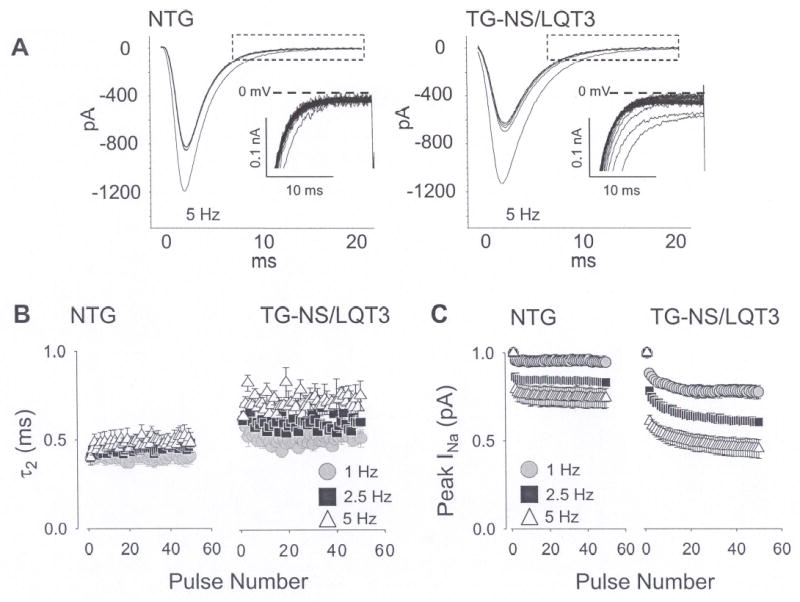
Rate-dependent reduction in peak and late INa. (A) A series of 50 consecutive INa currents are shown and these were elicited by 50 ms square steps from −80 to −20 mV at 5 Hz. Summaries for peak and late INa values at 1, 2.5, and 5 Hz are illustrated in the adjacent figures and these are plotted against pulse number. (B) Peak INa was measured as the peak current amplitude remaining relative to the 1st pulse. (C) INa,L was estimated quantitatively by fitting the declining phase of INa with a biexponential function, I/Imax = A1x(1-e(−t/τ1)) + A2x(1-e(−t/τ2)) and plotting the slow time component, τ2.
Gating defects of N1325S sodium channels in ventricular cells
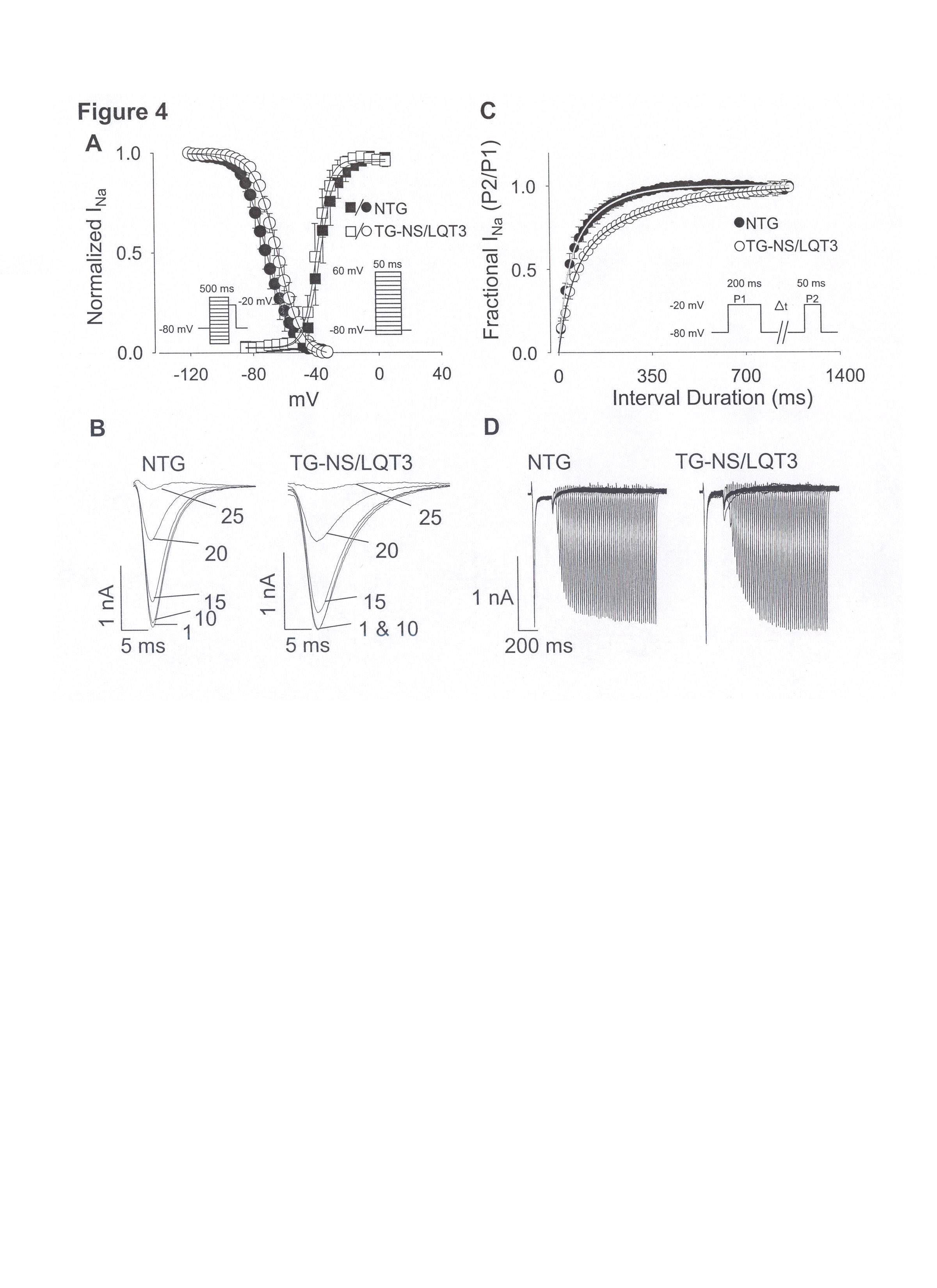
We and others carried out studies in Xenopus oocytes and HEK293 cells [11, 12] and showed that steady-state inactivation for N1325S channels was shifted to more positive potentials and that recovery from inactivation was slowed compared to wild type channels. In this study, we replicated the kinetic experiments on N1325S channels in native single ventricular cardiomyocytes. In Fig. 3A (supplementary data), no significant difference was observed for steady-state channel availability (i.e., activation curves) for opening between cells. We determined steady-state inactivation using a 500 ms condition pulse (to assess INa availability) followed by a test pulse at a fixed potential. Voltage-dependent current availability was altered in that steady-state half-inactivation was shifted 7 mV in the depolarizing direction (−63.89 ± 0.12 versus −71.03 ± 0.18 mV, P < 0.05; TG-NS/LQT3 and NTG cells, respectively). Representative INa traces (no. 1, 10, 15, 20, 25) elicited by the test pulse are illustrated in Fig. 3B and reflects this depolarizing shift in half-inactivation (increased INa availability) for TS-NS/LQT3 cells since peak INa at trace 1 and 10 are comparable in amplitude.
Fig. 3.
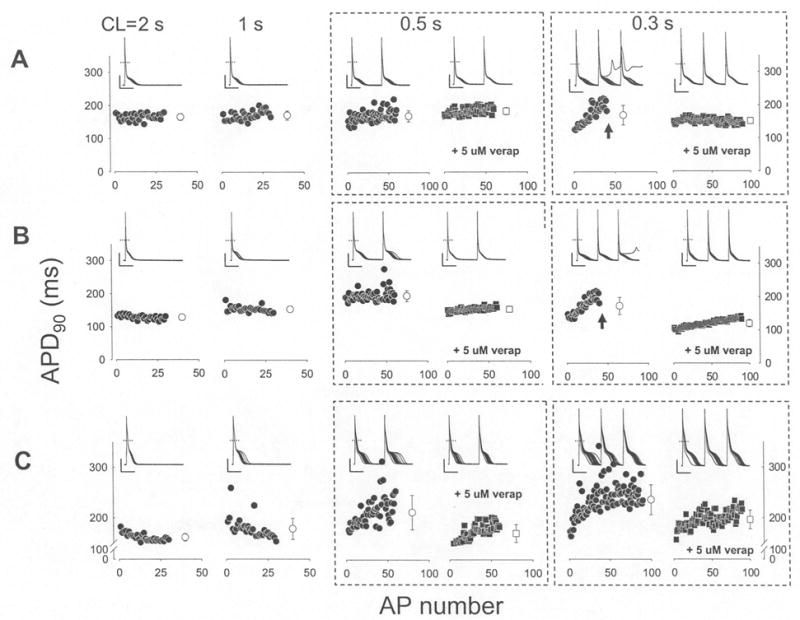
Verapamil prevents inhomogenous APD restitution at increasing rates. AP recordings were recorded from TG-NS/LQT3 cells at CLs of 2, 1, 0.5 and 0.3 sec. Representative recordings from three TG-NS/LQT3 cells (A, B, C) and their APD90 (90% repolarization) values (filled symbols) were plotted below as a function of AP number within each recording sweep; there was a total of 30 sweeps at each CL. An average value (open symbols ± SD) is indicated at the end of each data set. For CLs of 0.5 and 0.3 sec, recording traces and plots include pre- and post-administration of 5 uM verapamil. Failure to maintain stimulation capture (arrows) is indicated. The dashed horizontal line represents 0 mV and the vertical and horizontal solid lines indicate 20 pA and 0.1 sec scales, respectively.
Recovery from inactivation was assessed with a two-pulse protocol (Fig. 3C) in which the first pulse placed all channels in the inactivated state and the second pulse assessed the fraction of channels that had recovered from inactivation. The intervals are plotted on the abscissa of Fig. 3C and the averaged data for both cell groups were fitted with a biexponential function; representative traces are shown in Fig. 3D. The overall recovery times (τ1 and τ2) N1325S channels were slower than those for NTG channels. For these kinetic experiments, cells were held at −80 mV to better approximate channel kinetics at physiological conditions. These results in cardiomyocytes corroborate the earlier results in HEK293 cells [11] and oocytes [12] and indicate that in relation to NTG channels, N1325S channels are preferentially in the inactive channel state over the tested voltage range and are slow to recovery from inactivation. These alterations in channel inactivation are in accordance to the dispersed re-openings or increased window current reported to explain the late persistent sodium current [11, 12].
Action potential alternans and the effects of verapamil
Some LQTS patients with mutation N1325S died suddenly due to stress or excitement (non-bradycardia-induced trigger) and TG-NS/LQT3 mice developed VT during non-bradycardic conditions [3]. Thus, we hypothesized that a non-bradycardic arrhythmogenic mechanism was linked or associated with mutation N1325S. Our earlier studies showed that the action potential duration from TG-NS/LQT3 mocytes, but not those from NTG cells, became unstable and markedly prolonged with increased stimulation [3]. More specifically, the existence of AP alternans at decreasing diastolic intervals (Fig. 3, Table 1) led us to speculate that the generation and/or maintenance of AP prolongation were largely due to some instability in [Ca2+]i ions. Beat-to-beat variability of repolarization and abnormalities in [Ca2+]i in cardiomyocytes has been recognized [13–16]. As shown in Fig. 3 and Table 1, verapamil was effective in decreasing the incidence of TG-NS/LQT3 AP alternans and improving AP restitution. These results suggest that a reduction in Ca2+ influx provided assistance in the stabilization of the cardiac repolarization process. Whether this assistance was in the form of reducing the degree of [Ca2+]i inhomogeniety remains to be further elucidated. Reducing Ca2+ ion entry may be favorable in achieving intracellular ionic homeostasis given that late entry of Na+ ions may prompt TG-NS/LQT3 to extrude excess Na+ ions using the reverse mode of the Na/Ca exchanger at the risk of elevating [Ca2+]i levels. This may lead to more cytosolic Ca2+ and/or Ca2+ storage in the sarcoplasmic reticulum (SR), to which changes in SR Ca2+ content has been demonstrated to produced AP alternans [17]. Future experiments will be required to confirm this hypothesis.
Table 1.
APD90 values for NTG and TG-NS/LQT3 cells. APD values were calculated at 90% repolarization. A total of 30 sweeps was recorded at each CL and an average APD value ± SD was calculated. To clearly illustrate the beat-to-beat variability in TG-NS/LQT3 cells, these averages were listed for each TG cells. For NTG cells, the values were pooled together from a total of 8 cells and mean values ± SEM are listed. Cells that failed to maintain capture due to repolarization that exceeded the interpulse interval of the CL are indicated with “*”. TG-NS/LQT3 cells #1, 2, and 3 are the same representative cells used in Figure 5.
| Cycle Length (sec) | ||||||
|---|---|---|---|---|---|---|
| 2 | 1 |
0.5 |
0.3 |
|||
| −5 uM verap | +5 uM verap | −5 uM verap | +5 uM verap | |||
| NTG Cells | 44.6±2.6 | 46.3±3.3 | 46.9±5.9 | 48.6±4.1 | 47.3±3.1 | 50.6±2.3 |
| TG-NS/LQT3#1 | 165.1±10.1 | 170.4±13.6 | 168.6±16.5 | 175.5±10.2 | 172.4±28.9* | 151.6±7.3 |
| TG-NS/LQT3#2 | 127.8±6.4 | 152.5±8.5† | 194.5±16.5† | 133.5±4.9 | 172.7±25.8*† | 119.6±11.1 |
| TG-NS/LQT3#3 | 161.0±7.6 | 177.1±20.5† | 211.6±34.7† | 169.9±19.2 | 253.1±29.6† | 196.4±18.5† |
| TG-NS/LQT3#4 | 158.9±8.3 | 169.4±8.5† | 178.8±23.7† | 166.9±6.8 | 198.7±28.1† | 187.2±11.4† |
| TG-NS/LQT3#5 | 164.4±5.6 | 170.2±6.1 | 182.4±18.3† | 175.4±9.8† | 186.7±27.8† | 180.2±6.8† |
| TG-NS/LQT3#6 | 145.8±7.7 | 149.8±6.8 | 169.7±12.6† | 162.7±6.2† | 243.6±57.8*† | 167.1±8.6† |
indicates significance (P < 0.05) from values at CL = 2 sec.
We speculate that although INa,L is decreased at high stimulation rates, the already compromised Ca2+i-handling ability of TG-NS/LQT3 cells may result in a “mismanagement” of Ca2+i ion levels at shorter diastolic intervals as evident by the higher incidence and severity of AP alternans at shorter CLs. Poor restitution of APs during high stimulation rates has been previously reported elsewhere. In the ΔKPQ mouse model, a sudden increase in heart rate has been shown to induce a transient increase in APD and APD dispersion [18]. Patients with congenital LQTS (LQT1-3) exhibited T-wave alternans during sinus tachycardia [19, 20]. Increased APD dispersion has been identified as a proarrhythmic factor or mechanism involved during high stimulation rates. Evidence from a simulated LQTS study has suggested that EADs may occur at rapid heart rates due to abnormal calcium cycling [13]. The results from the present study provides a likely mechanistic explanation for non-bradycardia-induced LQT3 arrhythmogenesis associated with mutation N1325S. Whether the same mechanism applies to other LQTS mutations in SCN5A is another interesting question that should be studied on the mutation-by-mutation basis.
Conclusions
The results from this study demonstrate that one single mutation in the cardiac sodium channel gene SCN5A, N1325S, can result in both bradycardic- (26%) and non-bradycardic-induced (22%) cardiac events. The bradycardic events can be explained by our results that the contribution of INa,L is decreased with increasing stimulation rates (i.e., INa,L is more prevalent at slow stimulation rates). One potential mechanism for the non-bradycardic cause of cardiac events with mutation N1325S is an irregularity in [Ca2+]i homeostasis (particularly, at high stimulation rates) that may have resulted from cellular remodeling in response to the sodium channel mutation. Our results emphasize the complexity of genotype-phenotype correlation studies for LQTS mutations in SCN5A and the importance to study each mutation for detailed electrophysiological mechanisms.
Supplementary Material
{kind=link}
Acknowledgments
We thank Dr. G Kirsch for critical reading of the manuscript. This work was supported by the NIH grant R01 HL66251 (to QW), an AHA Established Investigator Award (to QW), and an AHA-Ohio-Affiliate Postdoctoral Fellowship (to SY).
References
- 1.Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA, Keating MT. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell. 1995;80:805–811. doi: 10.1016/0092-8674(95)90359-3. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Chen Q, Towbin JA. Genetics, molecular mechanisms and management of long QT syndrome. Ann Med. 1998;30:58–65. doi: 10.3109/07853899808999385. [DOI] [PubMed] [Google Scholar]
- 3.Tian XL, Yong SL, Wan X, Wu L, Chung MK, Tchou PJ, Rosenbaum DS, Van Wagoner DR, Kirsch GE, Wang Q. Mechanisms by which SCN5A mutation N1325S causes cardiac arrhythmias and sudden death in vivo. Cardiovasc Res. 2004;61:256–267. doi: 10.1016/j.cardiores.2003.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nuyens D, Stengl M, Dugarmaa S, Rossenbacker T, Compernolle V, Rudy Y, Smits JF, Flameng W, Clancy CE, Moons L, Vos MA, Dewerchin M, Benndorf K, Collen D, Carmeliet E, Carmeliet P. Abrupt rate accelerations or premature beats cause life-threatening arrhythmias in mice with long-QT3 syndrome. Nat Med. 2001;7:1021–1027. doi: 10.1038/nm0901-1021. [DOI] [PubMed] [Google Scholar]
- 5.Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantu F, Towbin JA, Keating MT, Hammoude H, Brown AM, Chen LS. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- 6.van den Berg MP, Haaksma J, Veeger NJ, Wilde AA. Diurnal variation of ventricular repolarization in a large family with LQT3-Brugada syndrome characterized by nocturnal sudden death. Heart Rhythm. 2006;3:290–295. doi: 10.1016/j.hrthm.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 7.Yong SL, Wang QK. Animal models for cardiac arrhythmias. In: Walker JM, Wang QK, editors. Methods in Molecular Medicine: Cardiovascular Disease. New Jersey: Humana Press; 2006. pp. 127–148. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Shen J, Li Z, Timothy K, Vincent GM, Priori SG, Schwartz PJ, Keating MT. Cardiac sodium channel mutations in patients with long QT syndrome, an inherited cardiac arrhythmia. Hum Mol Genet. 1995;4:1603–1607. doi: 10.1093/hmg/4.9.1603. [DOI] [PubMed] [Google Scholar]
- 9.Tan HL, Bezzina CR, Smits JP, Verkerk AO, Wilde AA. Genetic control of sodium channel function. Cardiovasc Res. 2003;57:961–973. doi: 10.1016/s0008-6363(02)00714-9. [DOI] [PubMed] [Google Scholar]
- 10.Nagatomo T, January CT, Ye B, Abe H, Nakashima Y, Makielski JC. Rate-dependent QT shortening mechanism for the LQT3 deltaKPQ mutant. Cardiovasc Res. 2002;54:624–629. doi: 10.1016/s0008-6363(02)00265-1. [DOI] [PubMed] [Google Scholar]
- 11.Wang DW, Yazawa K, George AL, Jr, Bennett PB. Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc Natl Acad Sci. 1996;93:13200–13205. doi: 10.1073/pnas.93.23.13200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, Kirsch GE. Multiple mechanisms of Na+ channel--linked long-QT syndrome. Circ Res. 1999;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 13.Huffaker R, Lamp ST, Weiss JN, Kogan B. Intracellular calcium cycling, early afterdepolarizations, and reentry in simulated long QT syndrome. Heart Rhythm. 2004;4:441–448. doi: 10.1016/j.hrthm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 14.Wan X, Laurita KR, Pruvot EJ, Rosenbaum DS. Molecular correlates of repolarization alternans in cardiac myocytes. J Mol Cell Cardiol. 2005;39:419–428. doi: 10.1016/j.yjmcc.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Goldhaber JI, Xie LH, Duong T, Motter C, Khuu K, Weiss JN. Action potential duration restitution and alternans in rabbit ventricular myocytes: the key role of intracellular calcium cycling. Circ Res. 2005;96:459–466. doi: 10.1161/01.RES.0000156891.66893.83. [DOI] [PubMed] [Google Scholar]
- 16.Pruvot EJ, Katra RP, Rosenbaum DS, Laurita KR. Role of calcium cycling versus restitution in the mechanism of repolarization alternans. Circ Res. 2004;94:1083–1090. doi: 10.1161/01.RES.0000125629.72053.95. [DOI] [PubMed] [Google Scholar]
- 17.Diaz ME, O’Neill SC, Eisner DA. Sarcoplasmic reticulum calcium content fluctuation is the key to cardiac alternans. Circ Res. 2004;94:650–656. doi: 10.1161/01.RES.0000119923.64774.72. [DOI] [PubMed] [Google Scholar]
- 18.Fabritz L, Kirchhof P, Franz MR, Nuyens D, Rossenbacker T, Ottenhof A, Haverkamp W, Breithardt G, Carmeliet E, Carmeliet P. Effect of pacing and mexiletine on dispersion of repolarisation and arrhythmias in DeltaKPQ SCN5A (long QT3) mice. Cardiovasc Res. 2003;57:1085–1093. doi: 10.1016/s0008-6363(02)00839-8. [DOI] [PubMed] [Google Scholar]
- 19.Cruz Filho FE, Maia IG, Fagundes ML, Barbosa RC, Alves PA, Sa RM, Boghossian SH, Ribeiro JC. Electrical behavior of T-wave polarity alternans in patients with congenital long QT syndrome. J Am Coll Cardiol. 2000;36:167–173. doi: 10.1016/s0735-1097(00)00694-x. [DOI] [PubMed] [Google Scholar]
- 20.Viskin S, Rosso R, Rogowski O, Belhassen B, Levitas A, Wagshal A, Katz A, Fourey D, Zeltser D, Oliva A, Pollevick GD, Antzelevitch C, Rozovski U. Provocation of sudden heart rate oscillation with adenosine exposes abnormal QT responses in patients with long QT syndrome: a bedside test for diagnosing long QT syndrome. Eur Heart J. 2004;4:469–475. doi: 10.1093/eurheartj/ehi460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.