

Abstract
We determined the sequence of the 152,372-bp genome of ϕYS40, a lytic tailed bacteriophage of Thermus thermophilus. The genome contains 170 putative open reading frames and three tRNA genes. Functions for 25% of ϕYS40 gene products were predicted on the basis of similarity to proteins of known function from diverse phages and bacteria. ϕYS40 encodes a cluster of proteins involved in nucleotide salvage, such as flavin-dependent thymidylate synthase, thymidylate kinase, ribonucleotide reductase, and deoxycytidylate deaminase, and in DNA replication, such as DNA primase, helicase, type A DNA polymerase, and predicted terminal protein involved in initiation of DNA synthesis. The structural genes of ϕYS40, most of which have no similarity to sequences in public databases, were identified by mass-spectrometric analysis of purified virions. Various ϕYS40 proteins have different phylogenetic neighbors, including Myovirus, Podovirus, and Siphovirus gene products, bacterial genes, and in one case, a dUTPase from a eukaryotic virus. ϕYS40 has apparently arisen through multiple acts of recombination between different phage genomes as well as through acquisition of bacterial genes.
Keywords: Thermus thermophilus, bacteriophage, genome, virion, proteomics, bioinformatics, DNA polymerase
Introduction
In the last decade, the genomes of several hundred phages have been completely sequenced (282 complete dsDNA phage genomes in the Genome Division of GenBank as of July 2006). While bacterial hosts of these phages are phylogenetically diverse, only ten of those completely sequenced phages are known to infect thermophilic microorganisms. Most of‘thermophilic’ phages were isolated from a small number of archaeal species 1-3. Sequence analysis revealed that archaeophages encode mostly uncharacterized proteins with no similarities to sequences in public databases, though more detailed examination revealed a limited number of recognizable ATPases, nucleotide salvage enzymes, and putative transcription factors 4. As of the time of this writing, the only sequenced genome of a phage from a thermophilic eubacterium is RM 378 that infects Rhodothermus marinus5.
During their development in a bacterial host, phages are known to regulate host macromolecular synthesis by modifying host transcription and translation machinery and making it serve the needs of the virus. Proteins from thermophilic bacteria are particularly amenable to structural studies of large complexes involved in DNA replication, DNA transcription, and RNA translation. Thus, structural and functional analysis of thermophilic phage-encoded regulators and their complexes with RNA polymerases, ribosomes, and other components of thermophilic bacteria can provide insights into molecular mechanisms of regulation of transcription, translation, and other cellular processes. With these ideas in mind, we determined the genomic sequence of ϕYS40, a large myophage hosted by the thermophilic bacterium Thermus thermophilus (temperature range from 56 to 78°C) 6. Here, we present the results of a preliminary study of the ϕYS40 genome and the proteome of ϕYS40 virions.
Results
Overview of the ϕYS40 genome
The sequence of the ϕYS40 genome was determined using the fimer technology and assembled into a single 152,372 bp contig using the phredPhrap package (see Materials and Methods). The G + C content of the ϕYS40 genome is 32.59%, which is significantly lower than that of its host (69.4%). Though the GC-content of ϕYS40 is close to values typical of the low-GC Gram-positive bacteria, there is no specific evolutionary affinity between sequences of ϕYS40 and these bacteria, and the GC-content of the phage may instead reflect specific aspects of phage molecular biology, for example distinct mutational bias of its DNA polymerase. ϕYS40 DNA appears to be unmodified as it is susceptible to digestion with all common methylation-sensitive restriction endonucleases tested (data not shown).
A total of 170 ORFs were predicted in the ϕYS40 genome (Table 1, Fig. 1). The intergenic regions were screened for additional genes by searching GenBank, GenPept, and the database of unfinished microbial genomes at NCBI, but no additional conserved ORFs were found. The predicted ϕYS40 ORFs are between 43 and 1744 codons in length. As with most other phages, the genome of ϕYS40 is tightly packed: coding sequences occupy 95% of the ϕYS40 genome. There are 46 cases of overlaps (from 1 to 40 bases long) between neighboring ORFs. The longest non-coding region (390 bp) lies between ORF138 and ORF139. Most of the 170 predicted ORFs start at the AUG codon, 22 ORFs use GUG codon, and three use UUG. At the ends of ϕYS40 genes, there are 90 TAA stop codons, 66 TGA codons, and 16 TAG codons.
Table 1.
Gene products of phage ϕYS40 and their predicted molecular functions.
| ORF name | ORF strand/positiona | ORF length (amino acids) | The best database match with validated similarity | Taxonomic origin of the best match | Function and other propertiesb |
|---|---|---|---|---|---|
| 1 | - / (7..1938) | 643 | 34419532 | Vibrio phage KVP40 | distal tail fiber protein |
| 2 | - / (1941..4586) | 881 | unknown | ||
| 3 | - / (4573..7410) | 945 | 48696430 | Staphylococcus phage K | portal protein |
| 4 | - / (7412..8068) | 218 | 90591438 | Flavobacterium johnsoniae UW101 | TM, unknown |
| 5 | - / (8096..8530) | 144 | 19924248 | Methanocaldococcus jannaschii | S-adenosylmethionine decarboxylase (adoMetDC) |
| 6 | - / (8564..8788) | 74 | unknown | ||
| 7 | - / (8801..9412) | 203 | unknown | ||
| 8 | - / (9399..9941) | 180 | 9631083 | Lymantria dispar nucleopolyhedrovirus | dUTPase |
| 9 | - / (9955..10782) | 275 | 33357605 | Thermotoga maritima | flavin-dependent thymidylate synthase |
| 10 | - / (10816..11331) | 171 | unknown | ||
| 11 | - / (11310..11783) | 157 | 33860394 | Burkholderia cepacia phage Bcep22 | gp18, unknown function |
| 12 | - / (11776..12795) | 339 | 23029929 | Microbulbifer degradans | RecA/RadA recombinase |
| 13 | - / (12792..13367) | 191 | 46200225 | Thermus thermophilus HB27 | Rad52 strand-exchange protein |
| 14 | - / (13413..14756) | 447 | 22978288 | Ralstonia metallidurans | DNA helicase DnaB |
| 15 | - / (14743..15036) | 97 | unknown | ||
| 16 | 15124..15453 | 109 | unknown | ||
| 17 | 15467..16576 | 369 | 23029305 | Microbulbifer degradans | IMP dehydrogenase / GMP reductase |
| 18 | 16640..17050 | 136 | 23110678 | Novosphingobium aromaticivorans | DNA binding HTH-domain protein, transcription regulator |
| 19 | - / (17108..18343) | 411 | Major structural protein | ||
| 20 | - / (18400..18837) | 145 | unknown | ||
| 21 | - / (18834..19214) | 126 | unknown | ||
| 22 | - / (19187..19960) | 257 | unknown | ||
| 23 | - / (19944..21620) | 558 | 27262500 | Heliobacillus mobilis | DNA primase bacterial DnaG type |
| 24 | - / (21669..22277) | 202 | 37526389 | Photorhabdus luminescens | thymidine kinase |
| 25 | - / (22302..23015) | 237 | 15595102 | Borrelia burgdorferi | ATP-dependent ClpP protease |
| 26 | - / (22975..23901) | 308 | 9964625 | Roseobacter phage SIO1 | RecB family exonuclease |
| 27 | - / (23898..25247) | 449 | 15900485 | Streptococcus pneumoniae | DEAD domain helicase |
| 28 | 25396..26796 | 466 | unknown | ||
| 29 | 26822..27331 | 169 | 52216967 | Bacteroides fragilis YCH46 | sugar-phosphate nucleotidyltransferas e |
| 30 | - / (27328..29085) | 585 | unknown | ||
| 31 | - / (29090..29803) | 237 | unknown | ||
| 32 | - / (29818..30291) | 157 | unknown | ||
| 33 | 30387..32498 | 703 | 29348669 | Bacteroides thetaiotaomicron | DNA polymerase, without N-terminal 5-3 exonuclease domain |
| 34 | - / (32491..32781) | 96 | 3 TMs, unknown | ||
| 35 | - / (32768..33034) | 88 | 2 TMs, unknown | ||
| 36 | - / (33031..33309) | 92 | unknown | ||
| 37 | 33381..33746 | 121 | unknown | ||
| 38 | 33730..34158 | 142 | 21229604 | Xanthomonas campestris | deoxycytidylate deaminase |
| 39 | 34188..34616 | 142 | unknown | ||
| 40 | 34631..35155 | 174 | unknown | ||
| 41 | 35201..37594 | 797 | 23104360 | Azotobacter vinelandii | ribonucleotide reductase, alpha subunit, the N-terminus |
| 42 | 37607..38206 | 199 | 20808702 | Thermoanaerobacter tengcongensis | ribonucleotide reductase, alpha subunit, the C-terminus |
| 43 | 38240..38446 | 68 | unknown | ||
| 44 | 38459..38911 | 150 | unknown | ||
| 45 | 38898..39227 | 109 | unknown | ||
| 46 | 39224..39439 | 71 | unknown | ||
| 47 | 39441..39884 | 147 | 4 TMs, unknown | ||
| 48 | 39877..40185 | 102 | unknown | ||
| 49 | 40201..40548 | 115 | unknown | ||
| 50 | 40558..41013 | 151 | unknown | ||
| 51 | 41010..42482 | 490 | unknown | ||
| 52 | 42536..43408 | 290 | 45914890 | Mesorhizobium sp. BNC1 | UDP-3-O-[3-hydroxy-myristory] glucosamine N-acyltransferase |
| 53 | 43411..43938 | 175 | unknown | ||
| 54 | 43940..44425 | 161 | unknown | ||
| 55 | - / (44426..45127) | 233 | 23055325 | Geobacter metallireducens | unknown |
| 56 | 45187..46209 | 340 | 51891857 | Symbiobacterium thermophilum | conserved bacterial protein, unknown |
| 57 | 46199..47536 | 466 | 42521856 | Bdellovibrio bacteriovorus | spore cortex synthesis protein SpoVR |
| 58 | 47564..49414 | 616 | unknown | ||
| 59 | 49453..51312 | 619 | 23112542 | Desulfitobacterium hafniense | putative serine protein kinase |
| 60 | 51410..51997 | 195 | 29366771 | Streptomyces phage phi-BT1 | putative dNMP kinase |
| 61 | 52035..52484 | 149 | |||
| 62 | - / (52477..54345) | 622 | 15668504 | Methanocaldococcus jannaschii | terminase large subunit |
| 63 | - / (54320..55108) | 262 | unknown | ||
| 64 | - / (55105..55485) | 126 | unknown | ||
| 65 | - / (55466..56017) | 183 | 22855150 | Bacillus phage B103 | terminal protein |
| 66 | 56049..56315 | 88 | unknown | ||
| 67 | - / (56362..57102) | 246 | unknown | ||
| 68 | - / (57104..57754) | 216 | unknown | ||
| 69 | - / (57775..59721) | 648 | 22973075 | Chloroflexus aurantiacus | tail sheath protein |
| 70 | - / (59782..60492) | 236 | unknown | ||
| 71 | - / (60495..61157) | 220 | 48696435 | Staphylococcus phage K | Zn ribbon, similar to archaeal transcription factor IIB |
| 72 | - / (61167..61682) | 171 | unknown | ||
| 73 | - / (61756..63168) | 470 | Major structural protein | ||
| 74 | - / (63204..64838) | 544 | 48696431 | Staphylococcus phage K | unknown, 3 coiled coil regions |
| 75 | - / (64838..65098) | 86 | unknown | ||
| 76 | - / (65085..69662) | 1525 | unknown | ||
| 77 | - / (69684..74918) | 1744 | unknown, 3 coiled coil regions | ||
| 78 | - / (74931..75296) | 121 | unknown | ||
| 79 | - / (75309..79883) | 1524 | 40744644 | Aspergillus nidulans | helicase (DEAD motif replaced by DDAE) |
| 80 | - / (79880..80743 | 287 | unknown | ||
| 81 | - / (80788..82740) | 650 | unknown | ||
| 82 | - / (82771..84609) | 612 | unknown | ||
| 83 | - / (84867..85094) | 75 | unknown | ||
| 84 | - / (85328..85558) | 76 | unknown | ||
| 85 | - / (85767..85919) | 50 | unknown | ||
| 86 | - / (86022..86273) | 83 | unknown | ||
| 87 | - / (86382..86618) | 78 | unknown | ||
| 88 | - / (86909..87154) | 81 | unknown | ||
| 89 | - / (87505..87990) | 161 | 15805515 | Deinococcus radiodurans | 2 TMs, unknown |
| 90 | - / (88074..88529) | 151 | unknown | ||
| 91 | - / (88642..89250) | 202 | unknown | ||
| 92 | - / (89349..89783) | 144 | unknown | ||
| 93 | - / (89796..90221) | 141 | unknown | ||
| 94 | - / (90481..90927) | 148 | unknown | ||
| 95 | - / (91036..91212) | 58 | unknown | ||
| 96 | 91231..91359 | 43 | unknown | ||
| 97 | - / (91417..91824) | 135 | 3 TMs, unknown | ||
| 98 | - / (91835..92380) | 181 | unknown | ||
| 99 | - / (92503..93045) | 180 | unknown | ||
| 100 | - / (93045..93635) | 196 | unknown | ||
| 101 | - / (93619..94131) | 170 | unknown | ||
| 102 | - / (94337..94873) | 178 | unknown | ||
| 103 | - / (94885..95373) | 162 | unknown | ||
| 104 | - / (95510..96025) | 171 | unknown | ||
| 105 | - / (96096..96626) | 176 | unknown | ||
| 106 | - / (96833..97354) | 173 | unknown | ||
| 107 | - / (97575..99263) | 562 | unknown | ||
| 108 | - / (99280..100323) | 347 | 15643692 | Thermotoga maritima | ATPase |
| 109 | - / (100462..101157) | 231 | unknown | ||
| 110 | - / (101227..101973) | 248 | unknown | ||
| 111 | - / (102138..102530) | 130 | unknown | ||
| 112 | - / (102531..103076) | 181 | unknown | ||
| 113 | - / (103077..103616) | 179 | unknown | ||
| 114 | - / (103616..104107) | 163 | 11992695 | Escherichia coli | glycosyltransferase |
| 115 | - / (104451..104693) | 80 | unknown | ||
| 116 | - / (104803..105279) | 158 | unknown | ||
| 117 | - / (105422..105979) | 185 | unknown | ||
| 118 | - / (105969..106520) | 183 | unknown | ||
| 119 | - / (106510..107076) | 188 | unknown | ||
| 120 | - / (107090..107539) | 149 | unknown | ||
| 121 | - / (107552..108046) | 164 | unknown | ||
| 122 | - / (108141..108644) | 167 | unknown | ||
| 123 | - / (108772..109290) | 172 | unknown | ||
| 124 | - / (109328..109819) | 163 | unknown | ||
| 125 | - / (109998..110513) | 171 | 18462664 | Shigella flexneri | unknown |
| 126 | - / (110561..111145) | 194 | unknown | ||
| 127 | - / (111157..111654) | 165 | unknown | ||
| 128 | - / (111663..112133) | 156 | unknown | ||
| 129 | - / (112165..112677) | 170 | unknown | ||
| 130 | - / (112689..113195) | 168 | unknown | ||
| 131 | - / (113202..113630) | 142 | unknown | ||
| 132 | 113852..114388 | 178 | coiled coil, unknown | ||
| 133 | - / (114385..115032) | 215 | unknown | ||
| 134 | - / (115155..115724) | 189 | unknown | ||
| 135 | - / (115727..116299) | 190 | unknown | ||
| 136 | - / (116271..116693) | 140 | unknown | ||
| 137 | 116815..117474 | 219 | unknown | ||
| 138 | - / (117442..118005) | 187 | unknown | ||
| 139 | - / (118395..119999) | 534 | 19552983 | Corynebacterium glutamicum | unknown |
| 140 | - / (120226..120777) | 183 | unknown | ||
| 141 | 120821..120994 | 58 | unknown | ||
| 142 | - / (120953..123997) | 1014 | coiled coil, unknown | ||
| 143 | - / (124012..124536) | 174 | unknown | ||
| 144 | - / (124553..125593) | 346 | 10956653 | Rhodococcus equi | M27/M37 peptidase |
| 145 | - / (125598..126548) | 316 | unknown | ||
| 146 | - / (126553..126813) | 86 | 3 TMs, unknown | ||
| 147 | 126870..127055 | 61 | unknown | ||
| 148 | 127065..127460 | 131 | unknown | ||
| 149 | 127471..127959 | 162 | unknown | ||
| 150 | 127979..129967 | 662 | 34762157 | Fusobacterium nucleatum | putative baseplate assembly protein |
| 151 | 129964..131859 | 631 | unknown | ||
| 152 | 131870..134260 | 796 | 903862 | Escherichia coli phage K3 | wac fibritin neck whisker |
| 153 | 134253..136364 | 703 | unknown | ||
| 154 | 136388..137287 | 299 | unknown | ||
| 155 | 137294..137644 | 116 | unknown | ||
| 156 | 137634..138497 | 287 | unknown | ||
| 157 | 138469..139269 | 266 | unknown | ||
| 158 | 139253..143296 | 1347 | unknown | ||
| 159 | 143322..143846 | 174 | unknown | ||
| 160 | 144155..144367 | 70 | unknown | ||
| 161 | - / (144357..145424) | 355 | 15674141 | Lactococcus lactis | Radical SAM superfamily enzyme |
| 162 | - / (145421..146374) | 317 | unknown | ||
| 163 | - / (146390..147022) | 210 | unknown | ||
| 164 | 147094..147639 | 181 | unknown | ||
| 165 | 147677..148306 | 209 | unknown | ||
| 166 | 148300..148689 | 129 | unknown | ||
| 167 | 148736..150229 | 497 | unknown | ||
| 168 | 150256..151341 | 361 | unknown | ||
| 169 | 151338..151907 | 189 | unknown | ||
| 170 | 151894..152157 | 87 | unknown |
YS40 virion proteins detected by MudPIT are indicated in red.
position of the ORFs in the phage YS40 genome; “-” indicates a leftwards transcription orientation.
presence of transmembrane domains (TM) and coiled coil regions are indicated.
Figure 1.
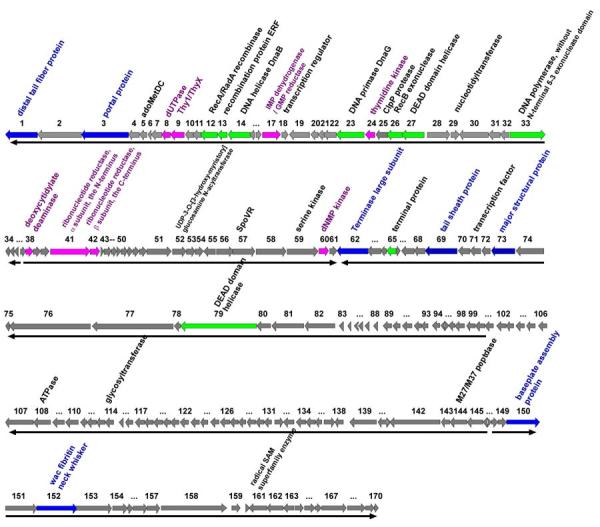
The ϕYS40 genome.
Bacteriophage ϕYS40 genome is schematically presented with predicted ORFs indicated by arrows. Arrow direction indicates the direction of transcription. Several ORFs with clear functional predictions for their products are color-coded (see also Table 1 for more details).
Two-thirds of the ϕYS40 genes (114 genes) are transcribed in one direction, designated as leftward in the genome map (Fig. 1), and 56 genes are transcribed in the rightward direction. The G+C content is approximately the same for both sets of ORFs. Taking a set of genes transcribed in the same direction and having no more than three consecutive intruders (i.e., genes transcribed in a different direction) as a cluster, we find four gene clusters in the ϕYS40 genome. The ORF1-ORF36 and ORF62-ORF146 clusters are transcribed in the leftward direction, and ORF37-ORF61 and ORF147-ORF170 clusters are transcribed in the rightwards direction (Fig. 1). The probability of obtaining each of the four clusters by chance, calculated using equation 2 from Durand and Sankoff 7 is less than 0.1, indicating that at least part of the clustering may be due to evolutionary or functional constraints.
tRNA genes
Using the tRNA scan-SE program, we identified three tRNA genes in the ϕYS40 genome. The tRNA1 gene overlaps with ORF61, whereas the tRNA2 and tRNA3 genes are both located between ORF139 and ORF140. Other large tailed dsDNA bacteriophages, such as coliphage T4 8, vibriophage KVP40 9, and phage phiKZ of P. aeruginosa 10 also encode several tRNAs.
The ϕYS40 tRNA1 and tRNA3 recognize ACA (threonine) and AGA (arginine) codons, respectively. These codons, while overrepresented in the ϕYS40 genome, are the rarest threonine and arginine codons in T. thermophilus genes. tRNA2 has a CAU anticodon, which would correspond to methionine codon AUG if C34 in the wobble position is unmodified. In homologous tRNAs from a number of bacteria and bacteriophages, the corresponding cytidine is converted to lysidine, which results in the AUA (Ile) decoding 11-13. Determinants for tRNAIle identity are thought to consist of anticodon loop bases A37 and A38, the discriminator base A73, and conserved base pairs in the D-arm (U12·A23), the anticodon arm (C29·G41), and the acceptor arm (C4·G69)14. All these characteristics are present in ϕYS40 tRNA2, which therefore may decode the isoleucine codon AUA, another rare T. thermophilus codon that is much more frequent in ϕYS40 ORFs. Thus, ϕYS40-encoded tRNAs may ensure efficient decoding of codons that are overrepresented in the phage genome relative to its host.
Sequence analysis of predicted ϕYS40 proteins
Analysis of intrinsic features of protein sequences indicates that seven ϕYS40 ORFs encode proteins with putative transmembrane domains (from one to three) and four ϕYS40 proteins are predicted to have coiled-coil regions. Only one protein, gp107, is predicted to be strongly non-globular, and only one protein, gp35, contains an N-terminal secretion signal peptide. All deduced amino acid sequences were compared to proteins in the non-redundant database at NCBI using the PSI-BLAST program with a slightly relaxed cutoff for profile inclusion (-h parameter). The comparison showed that ∼25% of ϕYS40 proteins display sequence similarity to proteins of known function (Table 1).
ϕYS40 proteins involved in nucleotide metabolism
Like other large phage genomes, ϕYS40 encodes a number of enzymes involved in nucleotide metabolism. They are gp8, a homolog of mammalian/viral UTPase (EC 3.6.1.23); gp9, related to a predicted flavin-dependent thymidylate synthase (EC 2.1.1.148); GMP reductase gp17 (EC 1.7.1.7); thymidine kinase gp24 (EC 2.7.1.21); deoxycytidylate deaminase gp38 (EC 3.5.4.12); dNMP kinase gp60 (EC 2.7.4.-); and the catalytic α subunit of ribonucleotide reductase encoded by two adjoining ORFs, gp41 and gp42 (EC 1.17.4.1). Except for dUTPase gp8, all these gene products show stronger sequence similarity to prokaryotic or phage enzymes than to their eukaryotic or archaeal counterparts. The best database match and closest phylogenetic neighbor for dUTPase gp8 is dUTPase from Lymantria dispar nucleopolyhedrosis virus. Gene exchange between phages and bacteria has been suggested to account for odd gene phylogenies that are sometimes observed in the components of bacterial replication and transcription machinery 15. Our observation indicates that eukaryotic viruses, and perhaps their hosts, may also be involved in such exchange.
ϕYS40 proteins involved in DNA replication and recombination
ϕYS40 encodes most of the proteins required for replisome formation, namely gp14, a replication initiation helicase DnaB; gp23, a bacterial DnaG-family DNA primase; gp26, a RecB family exonuclease; gp33, a type A DNA polymerase, and gp27, a DEAD box helicase. Another predicted DEAD-box helicase is encoded by gp79. Based on the fact that gp79 is a part of the ϕYS40 virion, we suspect that it is involved in viral DNA packaging. ϕYS40 also encodes two recombination proteins, gp12, a RecA/RadA recombinase, and gp114, an ssDNA-annealing protein of the ERF family. There are no gene products with detectable sequence similarity to known ssDNA-binding proteins 16 or DNA ligases.
The product of gene 65 is of particular interest for understanding the replication mechanism of ϕYS40. It shows a striking sequence similarity to a portion of the terminal protein (TP) of B. subtilis phage ϕ29. The Ser232 residue of the TP protein forms a phosphoester bond with the 5'-terminal dAMP of the phage genome, and is essential for protein-primed replication of linear dsDNA genome of ϕ29 17-19. This serine is conserved in ϕYS40 gp65 (Fig. 2). Thus, it is likely that gp65 primes the replication of ϕYS40 genomic DNA. It should be noted that the ends of the ϕYS40 genome as presented in Fig. 1 are arbitrary, since no defined ends were revealed during genome sequencing and assembly, indicating that the ϕYS40 genome may be circularly permuted or may have direct terminal repeats. This matter requires further investigation.
Figure 2.
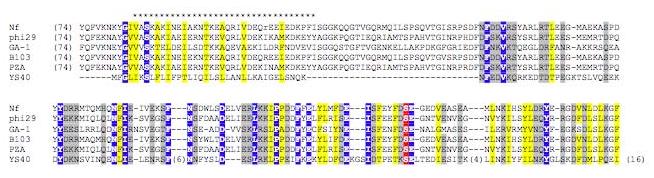
Sequence alignment of the TP proteins.
Multiple alignment of terminal proteins (TP) from ϕ29 family phages and phage ϕYS40 gp65. The stretch of * indicates a region of a predicted amphipathic alpha-helix in TP. Distances, in amino acid residues, from the ends of each sequence and between blocks, are shown in parentheses. A white font in blue indicates the residue identical in all sequences compared, yellow shading indicates the conservation of hydrophobic residues, grey shading indicates the conservation of polar and charged residues. The white font in red indicates the Ser232 that is essential for TP priming activity.
Properties of the ϕYS40 DNA polymerase
The ϕYS40 gp33 is a type A DNA polymerase, which contains a conserved nucleotidyltransferase domain and a 3'-5' exonuclease domain, but lacks the 5'→3' exonuclease domain. Since gp33 is the first known example of a type A DNA polymerase from a thermophilic phage, we expressed recombinant gp33 in E. coli and studied its properties in vitro. At 60-65 °C, recombinant gp33 exhibited moderate polymerization activity and very strong 3'→ 5'exonuclease activity toward both single-stranded DNA and double-stranded DNA substrates, even in the presence of 1 mM dNTP. As a result, at pH > 8.0 and low salt concentrations, the enzyme mostly hydrolyzed the primer. The increase of salt concentration partially inhibited the exonucleolytic activity and allowed primer elongation, until further increase inhibited the polymerase activity as well. The decay of primer-template substrate by gp33 exonuclease was abolished when primers were protected with thiolate modification, but the interference of the exonucleolytic activity during elongation resulted in poor DNA yield.
Gp33 was moderately thermostable. Both polymerase and exonuclease functions were lost after a 3-min incubation at 85 °C. At 75 °C, the polymerase activity decreased faster than the exonuclease activity; as a result, the enzyme produced shorter elongation products after heating. Similarly low thermostability has been reported for type B DNA polymerase from the Rhodothermus marinus phage (a half-life of 2 min at 90 °C 5). These observations indicate that both processivity of ϕYS40 DNA polymerase and its stability at elevated temperatures must be conferred by its interactions with other components of the replicative complex, in marked contrast with other DNA polymerases of bacteria and archaea, such as Taq or Pfu, which are processive and thermostable in the absence of cofactors.
Protein composition of ϕYS40 virions
To identify ϕYS40 structural proteins, ϕYS40 virions were purified by double sedimentation in CsCl gradients. The results of SDS-PAGE analysis of purified ϕYS40 virions are shown in Fig. 3. The two major protein components of the virion were identified by mass spectrometry as gp73 and gp19 (Fig. 3). These proteins may correspond to major head and tail proteins, but their function could not have been predicted by sequence comparison because of lack of database homologs.
Figure 3.
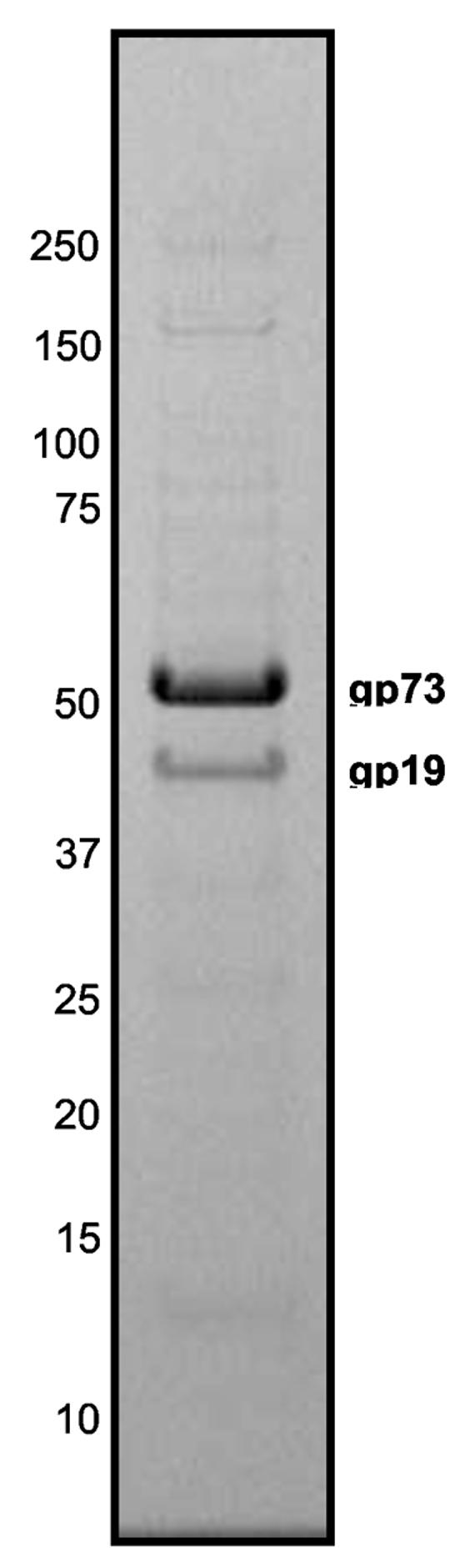
SDS-PAGE analysis of the ϕYS40 virion proteins.
The SDS gel shows the protein composition of purified ϕYS40 virions. The two major bands identified by mass-spectrometry are indicated.
Three independent ϕYS40 lysates of increasing titer (from 2×107 to 2×109 pfu/ml) were also directly examined by multidimensional protein identification technology, MudPIT 20 a shotgun proteomics approach where proteolytic peptides of a protein complex under study (in our case, phage virions) are generated, loaded onto triphasic microcapillary columns, eluted over several chromatography steps and analyzed directly by tandem mass spectrometry. Peptides matching 33 ϕYS40 proteins were detected in one or more of these samples. There were also 79 host proteins, all of which decreased in abundance when the lysates of higher titer were used as a starting material for CsCl purification (Supplementary Table A). In contrast, the NSAF (Normalized Spectral Abundance Factor, see Materials and Methods) values for ϕYS40 proteins increased with the titer of phage in the starting sample (Supplementary Table B). Gp73 and gp19 were detected at the highest levels in all three analyses in agreement with these being major structural proteins. With the exception of gp52 (annotated as a UDP-3-O-[3-hydroxymyristory] glucosamine N-acyltransferase), gp69 (tail sheath protein), gp79 (DEAD-Box helicase), gp150 (putative baseplate assembly protein), and gp152 (fibritin neck whisker), most ϕYS40 virion proteins identified in this analysis are novel proteins without any detectable database homologs. Interestingly, all multiply detected ϕYS40 virion proteins are the products of adjacent co-transcribed genes, except for ORF19 (Fig. 4C). In particular, a group of 13 proteins detected at high levels are the products of genes at the end of the largest cluster of ϕYS40 genes (ORF62-ORF146, above) that therefore may correspond to the late gene cluster.
Figure 4.
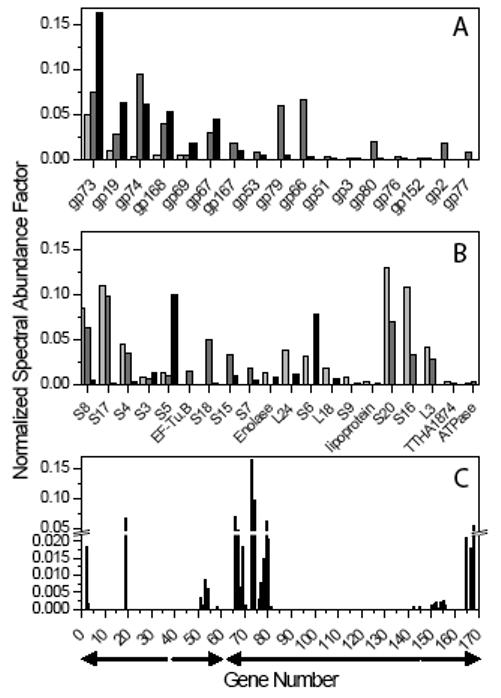
MudPIT analysis of ϕYS40 lysates.
- Normalized Spectral Abundance Factor (NSAF) values measured for ϕYS40 proteins detected in at least two of the three runs.
- NSAFs for contaminating T. thermophilus proteins detected in at least two of the three runs.
- All 33 ϕYS40 genes for which products were detected are plotted along the genome as a function of the measured NSAF values (when proteins were identified in several runs, maximal NSAF values are reported). The arrows under the x axis represent the position of the leftward and rightward predicted transcription clusters.
DISCUSSION
Bacteriophages may be the most abundant living entities on Earth. It has been proposed that the origin of dsDNA bacteriophages is as ancient as DNA replication itself and that the analysis of the currently known bacteriophages may provide clues to early evolution of cellular and viral genomes 15.
Here, we report a preliminary analysis of Thermus thermophilus bacteriophage ϕYS40 genome. The analysis shows that ϕYS40 does not easily fit into previously established groups of dsDNA bacterial viruses and may represent a distinct branch of the Myoviridae family. A substantial fraction of ϕYS40 genes codes for predicted proteins to which no function can be assigned; however, 25% of the ϕYS40-encoded proteins show detectable homology to their counterparts in a broad phylogenetic range of microorganisms, and some proteins are homologous to proteins found in other dsDNA bacteriophages infecting diverse hosts, such as Staphylococcus, Rhodothermus marinus, and Vibrio parahaemolyticus. In agreement with morphological data, predicted tail genes are mostly Myoviridae-related. Most of other ϕYS40 genes that have database homologs are, however, closer to either podoviral or siphoviral gene products: for instance, gp26 (RecB family exonuclease) and gp60 (dNMP kinase) are most closely related to homologs from a podovirus SIO1 and a λ-like siphovirus phi-BT1, respectively. Yet other genes are phylogenetically close to bacterial genes, and, in one case, to a homolog from a eukaryotic baculovirus. ϕYS40 has apparently arisen through multiple acts of recombination between different groups of phages and perhaps even their hosts.
Molecular adaptations to thermophily in various species are of great interest. Comparative studies of the genomes of thermophilic, hyperthermophilic, and mesophilic prokaryotes have suggested several attributes of thermostability at the levels of amino acid sequence, properties of folded proteins, and gene content. The proposed sequence level predictors of thermostability, such as large charged-versus-polar (CvP) amino acid ratio or (E + K)/(Q + H) ratio, are not conclusive in the case of ϕYS40, and genes that are indicative of the host ability to survive at extreme temperatures 21are missing from the ϕYS40 genome. Moreover, only seven ϕYS40 gene products have closest phylogenetic neighbors in thermophilic microorganisms.
In its genome size, ϕYS40 is similar to T4, an E. coli phage that is known to rely on host RNA polymerase for expression of its genes. During its development, T4 sequentially modifies host RNA polymerase to shut off transcription of host genes and to ensure correct expression of several classes of its own genes (reviewed in Ref. 22). Like T4, ϕYS40 does not encode its own RNA polymerase and therefore has to rely on the host enzyme for transcription of its DNA. The early genes of ϕYS40 should therefore be transcribed by the T. thermophilus RNA polymerase holoenzyme, most likely containing general initiation factor σA. Preliminary analysis reveals the presence of sequences with strong similarities to bacterial housekeeping sigma promoters in front of many ϕYS40 genes, but no such sequences are found in front of genes coding for ϕYS40 structural proteins (A. Sevostyanova, M. Gelfand and KS, unpublished observations). Structural genes, which should be expressed late in infection, must be therefore transcribed by a modified form of host RNA polymerase. Further biochemical studies may reveal ϕYS40 proteins that are required for these modifications.
MATERIALS AND METHODS
Cell growth and phage infection
The bacterial strain Thermus thermophilus HB8 and ϕYS40 were generously provided by Dr. Tairo Oshima, Tokyo University of Pharmacy & Life Science. The cells and phage were grown overnight in the Tth medium (0.8% polypeptone, 0.4% yeast extract, 0.2% NaCl, and 0.35 M CaCl2 and 0.4 M MgSO4) at 65°C with vigorous agitation.
To isolate individual ϕYS40 plaques, 1 ml of overnight HB8 culture (OD600∼1.6) was centrifuged and resuspended in 100 μl of the Tth medium and combined with 5 μl dilutions of ϕYS40 stock, incubated for 15 min at 65°C, plated in soft Tth agar (0.7 %), and incubated overnight at 65°C. An individual plaque was picked up and subjected to two more rounds of plaque purification, before making a phage lysate stock solution. To this end, a single plaque was resuspended in a small volume of the Tth medium and mixed with 0.1 ml of overnight HB8 culture. The mixture was incubated for 15 minutes at 65 °C to allow phage absorption, 5 ml of fresh Tth medium was added and the culture was incubated on a rotary shaker at 65 °C until complete lysis occurred (usually overnight). Cell debris was removed from the lysate by centrifugation at 12,000g for 15 minutes. The resultant phage stock (6×109 pfu/ml) was saturated with chloroform and stored at 4 °C. The ϕYS40 stock was used to prepare larger amounts of phage lysate using a scale-up of the procedure described above.
Purification of ϕYS40 virions
DNase I and RNase A (each to a final concentration of 1 μg/ ml) were added to ϕYS40 lysed T. thermophilus culture followed by a 30-min incubation at 30°C. Solid NaCl was added a final concentration of 1 M and dissolved by swirling. The lysed culture was left on ice for 1 h and centrifuged at 11,000 g for 10 min at 4°C. To precipitate ϕYS40, PEG 8000 was added to the supernatant to the final concentration of 10% (w/v) followed by a 1-h incubation on ice. Precipitated ϕYS40 particles were recovered by centrifugation at 11,000g for 10 min at 4 °C. The phage pellet was resuspended in 2 ml of SM buffer (NaCl, MgSO4,Tris-HCl, pH7.5, 2% gelatin). The PEG 8000 and cell debris were extracted from the phage suspension by adding an equal volume of chloroform and centrifuged at 3,000g for 15 min at 4°C. 0.5 g of solid CsCl per milliliter of bacteriophage suspension was added to the aqueous phase, which contained the bacteriophage particles, and dissolved by gentle mixing. CsCl step gradients (three steps with 1.45, 1.50, and 1.70 g/l density) were performed in Beckman SW41 polypropylene centrifuge tubes at 22,000 rpm for 2 hrs at 4 °C and at 38.000 rpm for 24 hrs at 4 °C (Beckman SW50.1 rotor, Beckman Coulter, Fullerton, CA). Purified bacteriophage suspension was dialyzed twice at room temperature for 1 h against a 1000-fold volume of 10 mM NaCl, 50 mM Tris-HCl pH 8.0, 10 mM MgCl2.
Extraction of phage DNA
EDTA (to a final concentration of 20 mM), proteinase K (to a final concentration of 50 μg/ml), SDS (to a final concentration of 0.5%) were added to bacteriophage solution and incubated at 56°C for 1 h. An equal volume of phenol was added to chilled bacteriophage suspension, mixed, and centrifuged at 3000 g for 5 min at room temperature. The aqueous phase was extracted with a 50:50 mixture of equilibrated phenol and chloroform, and equal volume of chloroform. DNA was precipitated with ethanol.
Genome sequencing
Initial sequence data were obtained using mini shotgun library of phage DNA. Several rounds of sequencing reactions were performed directly on phage DNA using ThermoFidelase and Fimer technology23, 24. Trace assembly was done with phredPhrap package (http://www.phrap.com/) 25. The final round of sequencing resulted in one pseudocircular contig with a no- errors quality level.
Sequence analysis
ORFs of ϕYS40 were predicted using the GeneMark server (http://opal.biology.gatech.edu/GeneMark/heuristic_hmm2.cgi, Ref. 26). The PSI-BLAST program 27 was used to detect the homologs of ϕYS40 genes in the DNA and protein databases, with profile inclusion cutoff E-value in PSI-BLAST (-h parameter) set at 0.02. Both options for low-complexity filtering (-F parameter) and composition-based statistics (-t parameter) were sometime adjusted for better detection in sequence similarities. Phylogenetic analysis was performed using the programs in the PHYLIP package.28
tRNA genes were searched by using the tRNAscan-SE program. 29 Searches for the presence of the transmembrane helices and coiled coil regions were done with the aid of the SEALS package. 30
MudPIT
Three independent virion lysates were prepared by double sedimentation in CsCl gradients and had phage titers of 2×107 pfu/ml, 4.2×108 pfu/ml and 2×109 pfu/ml. These lysates were treated with for 30 minutes at 37°C with 0.1U of benzonase (Sigma, St. Louis, MO), then precipitated in 20% trichloroacetic acid, 100mM Tris-HCl, ph 8.5, overnight at 4°C. The dried protein pellets were denatured, reduced, alkylated and digested with endoproteinase LysC and trypsin (both from Roche Applied Science, Indianapolis, IN) as described previously. 31 Peptide mixtures were pressure-loaded onto split-triphasic microcapillary columns, installed in-line with a Quaternary Agilent 1100 series HPLC pump coupled to Deca-XP ion trap tandem mass spectrometer (ThermoElectron, San Jose, CA) and analyzed via seven-step chromatography as described in Ref. 31.
The MS/MS datasets were searched using SEQUEST 32 against a database of 171 YS40 predicted gene products, combined with 2224 protein sequences from Thermus thermophilus, strain HB8 (chromosome and large plasmid) downloaded from NCBI on 2005-08-01, as well as usual contaminants such as human keratins, IgGs, and proteases. In addition, to estimate background correlations, each sequence in the database was randomized (keeping the same amino acid composition and length) and the resulting “shuffled” sequences were concatenated to the “normal” sequences and searched at the same time (the total number of sequences searched was 5144).
DTASelect/CONTRAST program33 was used to select spectra/peptide matches with normalized difference in cross-correlation score (DeltCn) of at least 0.11, a minimum cross-correlation score (XCorr) of 1.8 for singly-, 2.5 for doubly-, and 3.5 for triply-charged spectra, a maximum Sp rank of 10, and a minimal length of 7 amino acids. In addition, the peptides had to be fully tryptic. No peptides matching shuffled protein sequences passed this criteria set. Spectral counts are considered to be a good estimation of absolute protein abundance34. To account for the fact that larger proteins tend to contribute more peptide/spectra, spectral counts are divided by protein length defining a Spectral Abundance Factor (SAF).35 SAF values are normalized against the sum of all SAFs for each run (removing redundant proteins) allowing us to compare protein levels across different runs using the Normalized Spectral Abundance Factor (NSAF) value.
ϕYS40 DNA polymerase
The gene encoding ϕYS40 DNA polymerase was PCR amplified using appropriate primers annealing at the beginning and the end of ϕYS40 gene 33 and containing engineered NdeI site CATATG overlapping with the initiating ATG codon of gene 33 and a HindIII site downstream of the termination codon (primer sequences are available from the authors upon request). The amplified fragment with treated with NdeI and HindIII and cloned into appropriately digested pet21d plasmid and transformed into the E. coli expression strain BL-21 pLysS. Cells were grown in 1 L of LB medium and induced with 1 mM IPTG. Cell pellet was dissolved in 15 ml of lysis buffer and centrifuged at 17000 rpm for 30 min (no heat treatment). Lysate was diluted to 0.25M NaCl, and applied on a Heparin Sepharose High-Trap column (GE Healthcare, Newark, NJ), equilibrated with 50 mM Tris pH 7.5, containing 0.25 M NaCl and 2 mM mercaptoethanol. After washing with the same buffer, ϕYS40 DNA polymerase was eluted in about 0.3-0.35 M NaCl and appeared to be over 80% pure by SDS-PAGE. Assays of its enzymatic activities were done essentially as described by Pavlov et al., Ref. 36.
Supplementary Material
Acknowledgments
This work was supported by NIH RO1 grant GM64530 (to KS) and NIH GM61898 to Seth Darst. The authors thank Galina Glazko and Frank Emmert-Streib (both from Stowers Institute) for assistance on the gene clustering analysis and the analysis on codon usage, respectively.
References
- 1.Palm P, Schleper C, Grampp B, Yeats S, McWilliam P, Reiter WD, Zillig W. Complete nucleotide sequence of the virus SSV1 of the archaebacterium Sulfolobus shibatae. Virology. 1991;185:242–250. doi: 10.1016/0042-6822(91)90771-3. [DOI] [PubMed] [Google Scholar]
- 2.Arnold HP, Zillig W, Ziese U, Holz I, Crosby M, Utterback T, Weidmann JF, Umayam LA, Teffera K, Kristjansson JK, Klenk HP, Nelson KE, Fraser CM. A novel lipothrixvirus, SIFV, of the extremely thermophilic crenarchaeon Sulfolobus. Virology. 2000;267:252–266. doi: 10.1006/viro.1999.0105. [DOI] [PubMed] [Google Scholar]
- 3.Wiedenheft B, Stedman K, Roberto F, Willits D, Gleske AK, Zoeller L, Snyder J, Douglas T, Young M. Comparative genomic analysis of hyperthermophilic archaeal Fuselloviridae viruses. J. Virol. 2004;78:1954–1961. doi: 10.1128/JVI.78.4.1954-1961.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prangishvili D, Garrett RA, Koonin EV. Evolutionary genomics of archaeal viruses: Unique viral genomes in the third domain of life. Virus Res. 2006;117:52–67. doi: 10.1016/j.virusres.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Hjorleifsdottir S, Hreggvidsson GO, Fridjonsson OH, Aevarsson A, Kristjansson JK. Bacteriophage RM 378 of a thermophilic host organism. Patent: WO 0075335-A 14-DEC-2000 Decode Genetics EHF. 2000
- 6.Sakaki Y, Oshima T. Isolation and characterization of a bacteriophage infectious to an extreme thermophile, Thermus thermophilus HB8. J. Virol. 1975;15:1449–1453. doi: 10.1128/jvi.15.6.1449-1453.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Durand D, Sankoff D. Tests for gene clustering. J. Comput. Biol. 2003;10:453–482. doi: 10.1089/10665270360688129. [DOI] [PubMed] [Google Scholar]
- 8.Miller ES, Kutter EM, Mosig G, Arisaka F, Kunisawa T, Rüger W. Bacteriophage T4 genome. Microbiol. Mol. Biol. Rev. 2003;67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller ES, Heidelberg JF, Eisen JA, Nelson WC, Durkin AS, Ciecko A, Feldblyum TV, White O, Paulsen IT, Nierman WC, Lee J, Szczypinski B, Fraser CM. Complete genome sequence of the broad-host-range vibriophage KVP40: comparative genomics of a T4-related bacteriophage. J. Bacteriol. 2003;185:5220–5233. doi: 10.1128/JB.185.17.5220-5233.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mesyanzhinov VV, Robben J, Grymonprez B, Kostyuchenko VA, Bourkaltseva MV, Sykilinda NN, Krylov VN, Volckaert G. The genome of bacteriophage ϕKZ of Pseudomonas aeruginosa. J. Mol. Biol. 2002;317:1–19. doi: 10.1006/jmbi.2001.5396. [DOI] [PubMed] [Google Scholar]
- 11.Matsugi J, Murao K, Ishikura H. Characterization of a B. subtilis minor isoleucine tRNA deduced from tDNA having a methionine anticodon CAT. J. Biochem. (Tokyo) 1996;119:811–816. doi: 10.1093/oxfordjournals.jbchem.a021312. [DOI] [PubMed] [Google Scholar]
- 12.Muramatsu T, Nishikawa K, Nemoto F, Kuchino Y, Nishimura S, Miyazawa T, Yokoyama S. Codon and amino-acid specificities of a transfer RNA are both converted by a single post-transcriptional modification. Nature. 1988;336:179–181. doi: 10.1038/336179a0. [DOI] [PubMed] [Google Scholar]
- 13.Muramatsu T, Yokoyama S, Horie N, Matsuda A, Ueda T, Yamaizumi Z, Kuchino Y, Nishimura S, Miyazawa T. A novel lysine-substituted nucleoside in the first position of the anticodon of minor isoleucine tRNA from Escherichia coli. J. Biol. Chem. 1988;263:9261–9267. doi: 10.1351/pac198961030573. [DOI] [PubMed] [Google Scholar]
- 14.Nureki O, Niimi T, Muramatsu T, Kanno H, Kohno T, Florentz C, Giege R, Yokoyama S. Molecular recognition of the identity-determinant set of isoleucine transfer RNA from Escherichia coli. J. Mol. Biol. 1994;236:710–724. doi: 10.1006/jmbi.1994.1184. [DOI] [PubMed] [Google Scholar]
- 15.Filée J, Forterre P, Laurent J. The role played by viruses in the evolution of their hosts: a view based on informational protein phylogenies. Res. Microbiol. 2003;154:237–243. doi: 10.1016/S0923-2508(03)00066-4. [DOI] [PubMed] [Google Scholar]
- 16.Ponomarev VA, Makarova KS, Aravind L, Koonin EV. Gene duplication with displacement and rearrangement: origin of the bacterial replication protein PriB from the single-stranded DNA-binding protein Ssb. J. Mol. Microbiol. Biotechnol. 2003;4:225–229. doi: 10.1159/000071074. [DOI] [PubMed] [Google Scholar]
- 17.Hermoso JM, Méndez E, Soriano F, Salas M. Location of the serine residue involved in the linkage between the terminal protein and the DNA of phage ϕ29. Nucleic Acids Res. 1985;13:7715–7728. doi: 10.1093/nar/13.21.7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garmendia C, Salas M, Hermoso JM. Site-directed mutagenesis in the DNA linking site of bacteriophage ϕ29 terminal protein: isolation and characterization of a Ser232----Thr mutant. Nucleic Acids Res. 1988;16:5727–5740. doi: 10.1093/nar/16.13.5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garmendia C, Hermoso JM, Salas M. Functional domain for priming activity in the phage ϕ29 terminal protein. Gene. 1990;88:73–79. doi: 10.1016/0378-1119(90)90061-u. [DOI] [PubMed] [Google Scholar]
- 20.Washburn MP, Wolters D, Yates JR., 3rd. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 21.Makarova KS, Wolf YI, Koonin EV. Potential genomic determinants of hyperthermophily. Trends Genet. 2003;19:172–176. doi: 10.1016/S0168-9525(03)00047-7. [DOI] [PubMed] [Google Scholar]
- 22.Nechaev S, Severinov K. Bacteriophage-induced modifications of host RNA polymerase. Annu. Rev. Microbiol. 2003;57:301–322. doi: 10.1146/annurev.micro.57.030502.090942. [DOI] [PubMed] [Google Scholar]
- 23.Slesarev AI, Mezhevaya KV, Makarova KS, Polushin NN, Shcherbinina OV, Shakhova VV, Belova GI, Aravind L, Natale DA, Rogozin IB, Tatusov RL, Wolf YI, Stetter KO, Malykh AG, Koonin EV, Kozyavkin SA. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4644–4649. doi: 10.1073/pnas.032671499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Polushin N, Malykh A, Morocho AM, Slesarev A, Kozyavkin S. High-throughput production of optimized primers (fimers) for whole-genome direct sequencing. Methods Mol. Biol. 2005;288:291–304. doi: 10.1385/1-59259-823-4:291. [DOI] [PubMed] [Google Scholar]
- 25.Ewing B, Hillier L, Wendl MC, Green P. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998;8:175–815. doi: 10.1101/gr.8.3.175. [DOI] [PubMed] [Google Scholar]
- 26.Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999;27:3911–392. doi: 10.1093/nar/27.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Altschul SF, Madden TI, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.6. Department of Genome Sciences, University of Washington; Seattle: 2005. Distributed by the author. [Google Scholar]
- 29.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Walker DR, Koonin EV. SEALS: a system for easy analysis of lots of sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1997;5:333–339. [PubMed] [Google Scholar]
- 31.Tomomori-Sato C, Sato S, Parmely TJ, Banks CA, Sorokina I, Florens L, Zybailov B, Washburn MP, Brower CS, Conaway RC, Conaway JW. A mammalian mediator subunit that shares properties with Saccharomyces cerevisiae mediator subunit Cse2. J. Biol. Chem. 2004;279:5846–5851. doi: 10.1074/jbc.M312523200. [DOI] [PubMed] [Google Scholar]
- 32.Eng J, McCormack AL, Yates JR., 3rd An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Amer. Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 33.Tabb DL, McDonald WH, Yates JR., 3rd. DTASelect and Contrast: Tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu H, Sadygov RG, Yates JR., 3rd. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem. 2004;76:4193–4201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 35.Powell DW, Weaver CM, Jennings JL, McAfee KJ, He Y, Weil PA, Link AJ. Cluster analysis of mass spectrometry data reveals a novel component of SAGA. Mol. Cell. Biol. 2004;24:7249–7259. doi: 10.1128/MCB.24.16.7249-7259.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavlov AR, Belova GI, Kozyavkin SA, Slesarev AI. Helix-hairpin-helix motifs confer salt resistance and processivity on chimeric DNA polymerases. Proc. Natl. Acad. Sci. U. S. A. 2002;99:13510–13515. doi: 10.1073/pnas.202127199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.