

Abstract
Polypeptide conformer interconversion in a low dielectric environment is shown to be highly dependent on water concentration. Water increases this rate by 103, apparently by catalyzing hydrogen bond exchange, and thereby presenting functional properties analogous to that of a foldase. This catalytic effect is demonstrated on the interconversion of a parallel gramicidin dimer into an antiparallel dimer. A Hill coefficient of 6.5 is observed, illustrating the highly cooperative nature of the process. Protein folding in nonpolar environs, such as the hydrophobic core of a protein or the hydrophobic domain of a lipid bilayer, may be contingent on and rate-limited by the scarcity of water.
The ubiquity of water belies the complexity of its biological roles. The insertion of water between turns in an α-helix is thought to represent intermediates in protein folding and unfolding (1). A partial monolayer of water has been shown to be necessary for enzymatic activity, presumably promoting surface dynamics essential for enzymatic activity (2). Barron and coworkers (3) have described water as the “lubricant of life,” facilitating conformational fluctuations through its hydrogen-bonding capacity. Wu and Gorenstein (4) also have described water as a “lubricant” that facilitates local dynamics. We previously have described a catalytic role for protic solvents mediating hydrogen bond exchange (5). Numerous interactions of water with protein surfaces and internal pockets have been documented with both x-ray crystallography (1, 6) and NMR (7, 8). Here the interactions of water on the surface of a polypeptide dimer catalyzes a structural rearrangement involving the breaking and reforming of hydrogen bonds. A kinetic analysis of this structural rearrangement is presented as a function of water concentration showing a true catalytic activity for water, analogous to foldase that catalyzes disulfide rearrangements in protein isomerization.
Protein folding occurs concomitantly with hydrogen bond exchange, a process difficult to accomplish in low dielectric environments. This realization has led to the recent finding of kinetically trapped polypeptide conformational states in lipid bilayer environments (9, 10) and to the hypothesis of protic solvent catalysis of hydrogen bond exchange (5). To investigate the role of water as a catalyst for hydrogen bond exchange and as a “foldase” in nonpolar environments, we have studied solvent-dependent conformational transitions of gramicidin A. This pentadecapeptide has alternating D and L amino acid residues, and both termini are blocked so that this polypeptide has no formal charges. In lipid bilayers the monovalent cation selective channel is formed by a symmetric single-stranded helical dimer (11). The structure is a β-strand in which all of the side chains are on one side of the strand, resulting from the alternating D/L stereochemistry, which forces the strand into a helix with parallel β-sheet-type hydrogen bonding. In addition, six antiparallel β-sheet hydrogen bonds form across the amino terminus to amino terminus junction at the center of the bilayer stabilizing the dimer. In organic solvents, such single-stranded dimers are not observed, but intertwined dimers either parallel or antiparallel with a left- or right-handed helical sense are formed (12, 13). Again, these structures are β-strand-type structures with β-sheet-type hydrogen bonding between the strands. In ethanol a mixture of four such double-stranded structures are present in equilibrium, exchanging on the few-second to few-minute time scale (13). Such kinetics result in distinct resonances in the solution NMR spectra, because the time scale is longer than the inverse of the chemical shift separation (in Hz) between resonances from identical residues in different conformers. Although the time frame for most protein folding events is on the millisecond to nanosecond time scale, the time frames for the conformational rearrangements of gramicidin A described here are from seconds to tens of megaseconds. This long time course is caused by a variety of factors and permits detailed kinetics to be uniquely elucidated by solution NMR methods. In less polar solvents than ethanol such as dioxane or benzene/ethanol (95:5 by volume) a single conformer, the antiparallel left-handed double helix (known as species 3) dominates at equilibrium (14–17).
METHODS
Gramicidin A was prepared by solid-phase peptide synthesis on an Applied Biosystems peptide synthesizer by using fluorenylmethoxycarbonyl blocking chemistry (18). Synthetic gramicidin when cleaved from the solid-phase support was more than 98% pure as judged by HPLC and was used without further purification. Nonpolar solvent, d8-Dioxane (CIL, Woburn, MA) was dried with a 3-Å molecular sieve for 48 hr (the molecular sieve was activated at 350°C before use). Sample preparation was completed in an argon atmosphere in a dry box. All sample tubes were immediately frozen in a liquid helium-filled dewar upon removing from the dry box and flame-sealed.
All NMR spectra were recorded on a Varian UnityPlus 720 MHz spectrometer at 30°C. The N-type data were collected for gradient-correlated spectroscopy (GCOSY) experiments with a gradient strength of 0.32 gauss/cm for a 1.9-ms duration time. GCOSY solution spectra were recorded with four scans (total experimental time 18 min) for data sets of 4,096 by 256 points. Kinetic monitoring for a sample typically was accomplished through an array of GCOSY experiments with appropriate time spacing between each experiment. NMR assignments were made by using total correlation spectroscopy, double quantum filtered correlated spectroscopy, and nuclear Overhauser effect spectroscopy experiments.
RESULTS AND DISCUSSION
Fig. 1 shows fingerprint (NH to CαH) regions of GCOSY spectra of gramicidin A in dioxane with and without water present. Fig. 1 C and F shows the 15 dominant cross peaks (red) from the antiparallel, species 3 conformation that represent the equilibrium conformation in this environment (dioxane with less than 1% water). Fig. 1A shows a spectrum that reflects the equilibrium population distribution of both parallel and antiparallel conformations formed in ethanol before dissolution in dioxane. Consequently, there are four or more sets of 15 cross peaks in this spectrum. In the presence of 90 water molecules per dimer (<1% by volume), the mixture of conformers in Fig. 1A converts to species 3 in less than 10 hr (Fig. 1C), whereas in the absence of water (≈ three residual waters per dimer) the conversion requires more than 1,500 hr (Fig. 1 D–F). Because the end-point distribution is essentially the same with and without water, the water is influencing the kinetic rate.
Figure 1.

The GCOSY fingerprint regions for gA (12 mM) solutions during structural rearrangement in the presence (A–C) and “absence” (D–F) of water. The red cross peaks in the spectra represent the backbone NH-CαH cross peaks of an antiparallel left-handed helical dimer (species 3), whereas the black cross peaks are from a mixture of parallel conformers. The horizontal axis is the actual time scale within which the NMR spectra were acquired and the vertical axis corresponds to the increase of the species 3 population over time. The initial and final peptide structures are shown as dimers with red/white double strands for the species 3 structure and black/white strands for the parallel structures. Trp15 at the peptide C terminus is displayed to emphasize the relative orientation of the two peptide strands.
The spectral intensities show complex behavior during the time course of this conformer rearrangement. At least three conformers are converting to one, but the pathway may be complex, with significant (i.e., spectrally visible) intermediates. Consequently, signals that are not present in Fig. 1A appear in the intermediate stages, such as Fig. 1D, and then vanish in Fig. 1F. The rate dependence on water concentration shown in Fig. 2A represents initial reaction rates observed by measuring the cross peak intensities for species 3 as a function of time. A very steep dependence on water concentration is observed, and although 60 water molecules per dimer have little effect on the rate, at 150 molecules per dimer the influence of water on the rate appears to be saturated. However, for these rapid rates the time resolution for observing initial kinetic rates by using the GCOSY data sets is poor and hence the error bars are large. In the presence of water the following equilibrium is rapidly achieved:
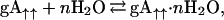 |
where gA↑↑ refers to a parallel gramicidin dimer and gA↑↑⋅nH2O refers to the hydrated parallel dimer. The weak dependence on water concentration below 60 water molecules per dimer suggests that n has to be a substantial number, before it can influence the kinetic rate significantly.
Figure 2.

(A) The initial conformational interconversion rates as a function of water concentration. The rate constants were measured from buildup curves of the fully assigned resonances for the antiparallel conformer, and they range from 1.4 ± 0.21 × 10−5 to 1.0 ± 0.2 × 10−2 min−1. The much increased error bars for the faster rates reflect the time required for acquiring the initial GCOSY data set for each sample (≈ 18 min). The kinetic results were fit by using Eq. 1. (B) The Hill plot with a slope of 6.5 indicates a very substantial cooperativity for the catalytic activity of water, suggesting that the breaking of one pair of peptide hydrogen bonds rapidly facilitates the subsequent disrupting of neighboring and next nearest neighboring hydrogen-bond pairs. The data corresponding to 3–95% kinetic rate saturation are used for calculating the maximal slope for the Hill coefficient.
When gramicidin is dissolved in the low dielectric mixture of benzene/ethanol (95:5 by volume) it has been shown that the ethanol binds to the indole NH and amide backbone sites as demonstrated by rotating-frame Overhauser effect spectroscopy cross peaks between the hydroxyl protons and the peptide (5). This observation has been used to explain how ethanol solvates gramicidin into a benzene environment. In the Fig. 3 Inset it is suggested that water as another protic solvent capable of hydrogen bonding is interacting with the backbone in much the same way. The minimum number of water molecules needed to satisfy an interaction with each amide is one water molecule for each hydrogen bond pair. The ideal model of the parallel double helix has 14 pairs of hydrogen bonds whereas the solution NMR structure of a parallel double helix in dioxane (19) provided evidence for just 12 pairs because of slight fraying of the helix at either end.
Figure 3.

A proposed free energy profile for water-catalyzed gramicidin conformational interconversion. (Inset) A parallel dimer model showing from this view six inter-strand hydrogen-bond pairs, each associated with an interacting water molecule. It is proposed here that these water molecules compete with and break the interpeptide hydrogen bonds, hence destabilizing the structure. In addition, water can substantially reduce the transition-state energy barrier by forming hydrogen bonds with those exposed polar backbone amide and carbonyl groups in nonpolar environments during the structural rearrangement. The free energies corresponding to the initial and transition states are labeled G↑↑ and G†↑↑, respectively. Note that the structure for the intermediate is drawn so as to suggest that one monomer has moved with respect to the other by a dipeptide. The subscripts +H2O/-H2O and black/purple lines denote the peptide system in the presence and absence of water, respectively. The interconversion rate enhancement is determined by ΔΔG = [(G†↑↑(+H2O) − G†↑↑(−H2O)) − (G↑↑(+H2O) − G↑↑(−H2O))], which reflects the additive effects of transition state stabilization and initial/final state destabilization.
To convert one double helix to another it has been suggested (20–22) that one monomer unscrews from the other in a series of steps that involves the breaking and reforming of hydrogen bonds after a translation and rotational motion of one monomer with respect to the other. Here, this model is further developed as a mechanism for this conformational interconversion. Because these are β-sheet-type structures the structural repeat unit is a dipeptide and hence this repeat unit represents the step size for the multistep conformational rearrangement. Therefore,
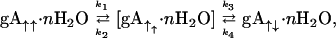 |
where gA↑↑⋅nH2O and gA↑↓⋅nH2O are the hydrated parallel and antiparallel double helices, respectively, each with 12–14 hydrogen bond pairs. A series of dipeptide plane-shifted intermediates with 10-, 8-, 6-, 4-, and 2-interpeptide hydrogen bond pairs are proposed and represented collectively by [gA↑↑⋅nH2O]. The formation of the first intermediate involves the cooperative destabilizing and breaking of the largest number of hydrogen bonds and is proposed to be the rate-limiting step characterized by k1 such that k2 ≫ k1. Similarly k3 ≫ k4, and, because of the greater stability of the antiparallel versus parallel structure in this solvent environment, k4 can be ignored in the kinetic analysis. The transition state for this rate-limiting step involves destabilization of each of the 12 or 14 hydrogen bond pairs and stabilization of the exposed amide and carbonyl groups (Fig. 3).
Classical Michaelis–Menten kinetics leads to
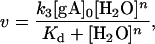 |
1 |
where v is the initial interconversion rate in units of min−1, Kd is a collection of rate constants and [gA]0 is total gramicidin concentration.
The data in Fig. 2A are fit with a solid line from the following result:
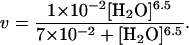 |
This analysis is further substantiated by rearranging Eq. 1, which also is known as the Hill equation (23).
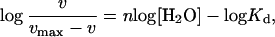 |
2 |
where vmax = k3[gA]0 and n is the Hill coefficient. The Hill plot presented in Fig. 2B is fit with the same kinetic analysis, leading to the observed Hill coefficient of 6.5. This large Hill coefficient suggests a very high degree of cooperativity in destabilizing and breaking hydrogen bonds in the rate-limiting step. Consequently, as one hydrogen bond pair is broken, neighboring and next nearest neighboring hydrogen bond pairs are broken in an essentially coherent process. This result represents substantial support for the conformational rearrangement model involving the unscrewing of one monomer from the other through coherent breaking and reforming of the hydrogen bonds between intertwined β-strands. An alternative model in which fraying of the ends of the polypeptide structure leads to conformational rearrangement would not require such cooperativity. These data provide evidence that many of the hydrogen bonds are breaking and reforming almost simultaneously.
The influence of water on a schematic potential energy surface for the peptide conformational interconversion is shown in Fig. 3. Water binds in the vicinity of each hydrogen bond pair, thereby destabilizing the dimer and raising the potential energy of the reactants and products (i.e., the ΔG of gA↑↑⋅nH2O and gA↑↓⋅nH2O). Furthermore, the water stabilizes the transition state involving broken hydrogen bonds that expose the significant partial charges of the amide groups in a low dielectric environment. In other words, the β-sheet-type hydrogen bonds are broken and water interacting with the polypeptide backbone is partially replacing this lost interaction energy. The important role of protic solvents in solvating and hence stabilizing initially broken hydrogen bonds is supported by an independent fluorescence study (24) on organic molecules. Benigo et al. proposed that hydrogen bond breaking involves an initial, partially solvated state very similar to our proposal here. Furthermore, their model describes a very high probability for reforming the same hydrogen bond from this partially solvated state. Here a relatively small motion on a screw axis of one chain with respect to the other can result in a new set of hydrogen bonds with only two fewer hydrogen bonds formed. The overall result in the present study is that the activation energy for the rate-limiting step is reduced substantially by the presence of water.
There are many important questions that remain about this mechanism (it is still very much a hypothesis), but this stepwise unscrewing of the dimer has gained considerable experimental support (21, 22, 25) as the mechanism occurring in a lipid environment where the double-stranded structure unwinds to the single-stranded channel state. Here, the data provide additional support for this mechanism of conformer interconversion in an organic solvent system. However, the most important conclusion from this study, albeit interpreted in light of this mechanism, but fundamentally independent of it, is that water has enhanced the kinetic rate of conformational interconversion without disturbing the conformer equilibrium. The mechanism presented simply illustrates how water could perform this activity.
The comparison of water with protein foldases is striking. Foldases, such as protein disulfide isomerase (PDI) can facilitate in vivo and in vitro protein folding by rearranging misplaced disulfide bonds through thio-disulfide exchange. Water facilitates hydrogen bond exchange. Foldases have a broad substrate specificity and no local sequence specificity and induce a moderate rate enhancement of 102 to 104 (26). Identical features can be described for water. Foldases, such as PDI are needed to correct misfolding steps in protein folding; similarly water may be used to correct misfolded or trapped off-pathway conformers in protein folding. Although such activity may be ubiquitous when water is abundant, its importance may be appreciated only in those environments where water is less common. In the hydrophobic core of a protein folding intermediate the residual water concentration may be a significant determinant of the structural rearrangement rate in the core region. The folding mechanism of membrane proteins is very complex (27); initial folding steps are assumed to take place in the bilayer hydrophobic/hydrophilic interface, a region of the membrane with a relatively high concentration of water. Once secondary and possibly tertiary and quaternary structural elements are folded the protein is inserted into the hydrophobic core of the bilayer. Although lipid bilayers are known to have a very low water concentration in their interstices, it is not known how much water a membrane protein folding intermediate brings into the bilayer to facilitate the final stages of protein folding. The need for a catalyst recently has been demonstrated by the observation that polypeptides can be kinetically trapped in nonminimum energy conformational states in a lipid bilayer (10, 11).
Acknowledgments
We thank Profs. Tim Logan and Don Caspar for helpful discussions, and we are indebted to the Florida State University and National High Magnetic Field Laboratory Facility staffs, especially J.B. Vaughn, Jr., N. Murali, and A. Blue. We also thank H. Henricks and U. Goli in the Bioanalytical Synthesis and Services Facility for their expertise and maintenance of the peptide synthesizer and HPLC equipment. This work has been supported by National Institutes of Health Grants GM49092 and AI23007, and the research was largely performed at the National High Magnetic Field Laboratory supported by National Science Foundation Cooperative Agreement DMR-9527035 and the State of Florida.
ABBREVIATION
- GCOSY
gradient-correlated spectroscopy
References
- 1.Sundaralingam M, Sekharudu Y C. Science. 1989;244:1333–1337. doi: 10.1126/science.2734612. [DOI] [PubMed] [Google Scholar]
- 2.Klibanov A M. Trends Biochem Sci. 1989;14:141–144. doi: 10.1016/0968-0004(89)90146-1. [DOI] [PubMed] [Google Scholar]
- 3.Barron L D, Hecht L, Wilson G. Biochemistry. 1997;36:13143–13147. doi: 10.1021/bi971323j. [DOI] [PubMed] [Google Scholar]
- 4.Wu J, Gorenstein D G. J Am Chem Soc. 1993;115:6843–6850. [Google Scholar]
- 5.Xu F, Wang A, Vaughn J B, Cross T A. J Am Chem Soc. 1996;118:9176–9177. [Google Scholar]
- 6.Yamano A, Heo N H, Teeter M M. J Biol Chem. 1997;272:9597–9600. doi: 10.1074/jbc.272.15.9597. [DOI] [PubMed] [Google Scholar]
- 7.Otting G, Liepinsh E, Wuthrich K. Science. 1991;254:974–980. doi: 10.1126/science.1948083. [DOI] [PubMed] [Google Scholar]
- 8.Ernst J, Clubb R, Zhou H, Gronenborn A, Clore G. Science. 1995;267:1813–1817. doi: 10.1126/science.7892604. [DOI] [PubMed] [Google Scholar]
- 9.Arumugam S, Pascal S, North C L, Hu W, Lee K-C, Cotten M, Ketchem R R, Xu F, Brenneman M, Kovacs F, et al. Proc Natl Acad Sci USA. 1996;93:5872–5876. doi: 10.1073/pnas.93.12.5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cotten M, Xu F, Cross T A. Biophys J. 1997;73:614–623. doi: 10.1016/S0006-3495(97)78097-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ketchem R R, Hu W, Cross T A. Science. 1993;261:1457–1460. doi: 10.1126/science.7690158. [DOI] [PubMed] [Google Scholar]
- 12.Urry D W, Glickson J D, Mayers D F, Haider J. Biochemistry. 1972;11:487–493. doi: 10.1021/bi00754a002. [DOI] [PubMed] [Google Scholar]
- 13.Veatch W R, Fossel E T, Blout E R. Biochemistry. 1974;13:5249–5256. doi: 10.1021/bi00723a001. [DOI] [PubMed] [Google Scholar]
- 14.Arseniev A S, Bystrov V F, Ivanov V T, Ovchinnikov Y A. FEBS Lett. 1984;165:51–56. [Google Scholar]
- 15.Bano M D, Braco L, Abad C. FEBS Lett. 1989;250:67–71. doi: 10.1016/0014-5793(89)80686-6. [DOI] [PubMed] [Google Scholar]
- 16.Pascal S M, Cross T A. J Biomol NMR. 1993;3:495–513. doi: 10.1007/BF00174606. [DOI] [PubMed] [Google Scholar]
- 17.Xu F, Cross T A. Magn Reson Chem. 1998;36:651–655. [Google Scholar]
- 18.Fields C G, Fields G B, Noble R L, Cross T A. Int J Pept Protein Res. 1989;33:298–303. doi: 10.1111/j.1399-3011.1989.tb01285.x. [DOI] [PubMed] [Google Scholar]
- 19.Pascal S M, Cross T A. J Mol Biol. 1992;226:1101–1109. doi: 10.1016/0022-2836(92)91055-t. [DOI] [PubMed] [Google Scholar]
- 20.Urry D W, Long M M, Jacobs M, Harris R D. Ann NY Acad Sci. 1975;264:203–220. doi: 10.1111/j.1749-6632.1975.tb31484.x. [DOI] [PubMed] [Google Scholar]
- 21.O’Connell A M, Koeppe R E II, Andersen O S. Science. 1990;250:1256–1259. doi: 10.1126/science.1700867. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Z, Pascal S M, Cross T A. Biochemistry. 1992;31:8822–8828. doi: 10.1021/bi00152a019. [DOI] [PubMed] [Google Scholar]
- 23.Fersht A. Enzyme Structure and Mechanism. New York: Freeman; 1977. pp. 272–273. [Google Scholar]
- 24.Benigo A J, Ahmed E, Mark B. J Chem Phys. 1996;104:7382–7394. [Google Scholar]
- 25.Cotten M, Fu R, Cross T A. Biophys J. 1999;76:1179–1189. doi: 10.1016/S0006-3495(99)77282-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Creighton T E. Protein Folding. New York: Freeman; 1992. pp. 480–488. [Google Scholar]
- 27.Engelman D M. Science. 1996;274:1850–1851. doi: 10.1126/science.274.5294.1850. [DOI] [PubMed] [Google Scholar]