

Abstract
Background: Although the adenoma to carcinoma pathway in colorectal cancer is well described, the mechanisms of carcinogenesis in the small intestine remain unclear.
Aims: The aim of this study was to investigate candidate genes in the genetic pathway of adenocarcinoma of the small intestine.
Subjects and methods: A total of 21 non-familial, non-ampullary adenocarcinomas of the small intestine were analysed. DNA was extracted from formalin fixed paraffin wax embedded tissue using standard techniques. The replication error (RER) status was determined by amplification of BAT26. The mutation cluster region (MCR) of the adenomatous polyposis coli (APC) gene was screened using polymerase chain reaction single strand conformational polymorphism and direct sequencing. Immunohistochemistry was performed on formalin fixed paraffin wax embedded tissue using monoclonal antibodies for hMLH1, hMSH2, β-catenin, E-cadherin, and p53.
Results: Fourteen male and seven female patients with a median age of 64 years (range 21–85) presented with adenocarcinoma of the duodenum (10), jejunum (7), and ileum (4). One cancer (5%) was found to be RER+, and all tumours stained positive for hMLH1 and hMSH2. No mutations were detected in the MCR of the APC gene. β-Catenin showed increased nuclear expression with loss of membranous staining in 10 cancers (48%). Absent or decreased membrane expression of E-cadherin was found in eight cancers (38%). Strong staining of p53 was found in the nucleus of five cancers (24%).
Conclusion: We did not detect mutations in the MCR of the APC gene, and this suggests that adenocarcinoma of the small intestine may follow a different genetic pathway to colorectal cancer. Abnormal expression of E-cadherin and β-catenin was common and reflects an early alternative to APC in this pathway in which mutations may be found in adenocarcinoma of the small intestine.
Keywords: small intestine, adenocarcinoma, replication error, adenomatous polyposis coli, β-catenin, E-cadherin, p53
Cancer of the small intestine is rare and accounts for only 1% of gastrointestinal malignancies,1 and this is despite the fact that the small intestine contains approximately 90% of the mucosal surface of the gastrointestinal tract.2 The incidence of colorectal cancer is 50 times that of small bowel cancer,3 yet the histopathological features of the two cancers are similar. Both cancers arise from adenomatous polyps4 and patients with one cancer are at increased risk of developing the other cancer.5
Adenocarcinoma together with carcinoid tumours are the most common histological types of cancer of the small intestine but patients may present with other primary malignant tumours, including lymphoma and leiomyosarcoma. Familial adenomatous polyposis (FAP) and Crohn's disease are recognised risk factors for small bowel cancer,4,6 and although adenocarcinomas cluster in the duodenum,6 the increased risk in Crohn's disease is mainly localised to the terminal ileum. Other risk factors include Peutz-Jeghers syndrome, coeliac disease, and hereditary non-polyposis colorectal cancer (HNPCC).7,8 It has been suggested that the rapid turnover of the small intestine mucosa, the relative absence of bacteria, and decreased transit time2,9 are all reasons why the incidence of cancers of the small intestine is less than that of the large intestine. Other suggestions include the alkaline pH within the small bowel, the liquid nature of small bowel contents, and a well developed local IgA mediated immune system.10
The diagnosis of adenocarcinoma of the small bowel is often delayed due to the patient having non-specific symptoms, and the subsequent difficulties that this produces in performing the relevant investigation.11 This undoubtedly contributes to the poor prognosis despite surgery and adjuvant chemotherapy and radiotherapy. Approximately 50% of adenocarcinomas of the small intestine are poorly differentiated, and the majority have infiltrated through the bowel wall at presentation.1
The histological progression to invasive colorectal cancer is termed “the adenoma-carcinoma sequence” and is accompanied by a series of well described genetic changes12,13 that involve activation of oncogenes and inactivation of tumour suppressor genes. Although the prevalence of cancer of the small intestine is low compared with colorectal cancer, the similarities in the two cancers suggest that they may share many of the genetic changes of carcinogenesis. If the two cancers differ in either the type of genetic changes or the frequency of these changes, then it may be hypothesised that the small bowel is resistant to the genetic events that occur in colorectal cancer. Furthermore, if a mechanism for any potential differences could be identified, this may have clinical applications in the future.
At present, there have been very few studies (usually with small numbers) of the molecular genetics of cancer of the small intestine. The aim of this study was to investigate the following candidate genes or their protein expression in the genetic pathway of adenocarcinoma of the small intestine: the mismatch repair genes hMLH1 and hMSH2, the adenomatous polyposis coli (APC) gene, β-catenin, E-cadherin, and p53.
HNPCC, which is characterised by microsatellite instability (or replication error, RER+) is caused by germline mutations in one of the DNA mismatch repair (MMR) genes.14 To date, inactivating mutations have been described in five mismatch repair genes: hMSH2, hMLH1, hPMS1, hPMS2, and GTBP (hMSH6).15–22 Although adenocarcinoma of the small intestine is 25 times more common in HNPCC kindreds than in the normal population,23 the incidence of RER+ sporadic adenocarcinoma of the small intestine is not known.
The APC gene, which is responsible for FAP, acquires truncating mutations in up to 80% of sporadic colorectal cancers.12 Unlike the germline mutations of FAP, 60% of mutations in sporadic colorectal cancer cluster in a region around codons 1200–1600—the mutation cluster region (MCR).24 This results in functions of the APC protein beyond the MCR being lost. This includes downregulation of the levels and activity of β-catenin,25,26 a 92 kDa protein which is important in the functional activities of both APC and E-cadherin. The selective advantage of APC mutations may be loss of regulation of β-catenin activity (which then accumulates in the nucleus), and this is supported by the finding of gain of function mutations of the β-catenin gene in those colorectal cancers with wild-type APC.27,28 E-cadherin encodes a 120 kDa transmembrane glycoprotein which localises mainly to the zonula adherens junctions and serves as the prime mediator of epithelial cell to cell adhesion. APC competes directly with E-cadherin for binding to β-catenin and therefore inactivation of either gene may be an indirect regulator of E-cadherin mediated adhesion. An alteration in any of these genes may cause abnormal architectural development of epithelium, which results in the initial development of adenomas in colorectal cancer.29
p53 is a nuclear oncosuppressor protein which is involved in the maintenance of genomic integrity. Mutations in the p53 gene occur commonly in a wide range of human cancers and result in increased expression of p53. The selective effect of p53 mutations is primarily on the frequency of programmed cell death or apoptosis.30 p53 mutations occur commonly in the adenoma to carcinoma sequence of colorectal cancer and are probably a late event.31
MATERIALS AND METHODS
Materials
Formalin fixed paraffin wax embedded archival tissue was available from a total of 21 non-familial, non-ampullary primary adenocarcinomas of the small intestine (table 1 ▶) (John Radcliffe Hospital, Oxford; University Hospital of Wales, Cardiff; Gloucestershire Royal Hospital, Gloucester; Southampton General Hospital, Southampton). Twelve male and nine female patients with a median age of 64 years (range 21–85) presented with adenocarcinoma of the duodenum (n=10, 48%), jejunum (n=7, 33%), and ileum (n=4, 19%). Six of the duodenal cancers were not resected and therefore only biopsy material was available for study. Of the remaining 15 cancers, there were three stage T4, 11 stage T3, and one stage T2 cancers. Three of the five cancers resected with lymph nodes had lymph node adenocarcinoma deposits. Fifteen cancers were moderately or well differentiated, with six being poorly differentiated.
Table 1.
Clinicopathological features of 21 non-familial, non-ampullary small bowel adenocarcinomas
| Immunohistochemistry | |||||||||||||
| Sex | Age (y) | Site | Stage | Differentiation | Lymph nodes | Mucinous | Vascular invasion | hMLH1 | hMSH2 | β-Catenin | E-cadherin | p53 | |
| 1 | M | 24 | Duodenum | T3N1 | Well | 2 of 8 | No | No | + | + | Reduced/cytoplasmic | + | + |
| 2 | F | 49 | Duodenum | T3Nx | Moderately | 0 | No | No | + | + | Reduced | + | + |
| 3 | M | 83 | Duodenum | Bx | Well | Bx | No | No | + | + | Reduced/nuclear | + | + |
| 4 | M | 53 | Jejunum | T3N1 | Poor | 2 of 7 | No | Yes | + | + | Reduced/nuclear | + | + |
| 5 | M | 72 | Jejunum | T3Nx | Well | 0 | No | Yes | + | + | Reduced/nuclear | − | − |
| 6 | F | 77 | Ileum | T3N0 | Moderately | 0 of 3 | No | No | + | + | Reduced/nuclear | − | − |
| 7 | M | 76 | Duodenum | Bx | Poor | Bx | Yes | Yes | + | + | Reduced | − | − |
| 8 | F | 85 | Duodenum | Bx | Well | Bx | No | No | + | + | Reduced/cytoplasmic | − | − |
| 9 | F | 77 | Jejunum | T4N1 | Poor | 2 of 4 | No | Yes | + | + | Reduced/nuclear | − | − |
| 10 | F | 54 | Duodenum | Bx | Moderately | Bx | No | No | + | + | Reduced/nuclear | − | − |
| 11 | M | 69 | Duodenum | Bx | Poor | Bx | No | No | + | + | Reduced/nuclear | − | − |
| 12 | F | 55 | Jejunum | T3N0 | Moderately | 0 of 1 | No | No | + | + | + | + | − |
| 13 | M | 69 | Jejunum | T3Nx | Moderately | 0 | No | Yes | + | + | Reduced/cytoplasmic | + | − |
| 14 | F | 82 | Ileum | T3Nx | Moderately | 0 | No | No | + | + | Reduced/nuclear | + | − |
| 15 | M | 72 | Ileum | T3Nx | Poor | 0 | No | Yes | + | + | + | + | + |
| 16 | M | 58 | Jejunum | T3Nx | Moderately | 0 | Yes | No | + | + | + | + | − |
| 17 | F | 50 | Duodenum | T2Nx | Well | 0 | No | Yes | + | + | + | + | − |
| 18 | F | 70 | Ileum | T3Nx | Poor | 0 | No | Yes | + | + | Reduced | − | − |
| 19 | M | 53 | Duodenum | Bx | Moderately | Bx | Yes | No | + | + | Reduced/nuclear | + | − |
| 20 | M | 58 | Ileum | T4Nx | Moderately | 0 | Yes | No | + | + | Reduced/nuclear | + | − |
| 21 | M | 21 | Duodenum | T4Nx | Moderately | 0 | No | No | + | + | Reduced/cytoplasmic | + | − |
DNA extraction
DNA was extracted from 15 μm paraffin sections using a standard Nucleon DNA extraction kit (SL-8502 Nucleon QC, Didsbury, Manchester, UK). Briefly, the paraffin sections were added to xylene at 37°C for 20 minutes, prior to rehydration with alcohol and water. Proteinase K solution was then added and left for 55°C for three hours. After chloroform extraction, genomic DNA was precipitated with ethanol.
Determination of the replication error (RER) status
In order to determine the RER status of these cancers, we amplified BAT-26, a single poly (A) tract, previously shown to be highly sensitive and specific for microsatellite instability,32 using fluorescent labelled primers and similar polymerase chain reaction (PCR) conditions to those previously described. PCR products were loaded on a 377 prism sequencer (ABI, Warrington, Cheshire, UK). Results were analysed using GeneScan software (version 2.0.2). All PCRs and analyses were repeated at least in duplicate. Any tumours presenting ambiguous results were further investigated using BAT-25.
Screening for mutations in the APC gene
Using previously described primers for exon 15E-I of the APC gene,33 the MCR was amplified specifically from genomic DNA. All reactions contained approximately 100 ng of template DNA in a total volume of 50 μl with final reaction concentrations of 1× PCR buffer (10 mM Tris/HCl pH 8.3, 50 mM KCl, 0.1% Triton, 3 mM MgCl2), 200 mM dNTPs, 0.2 mM of each primer, and 1 U of Taq polymerase. Amplification was performed using a protocol of 95°C for five minutes, 35 cycles of 95°C for one minute, 60°C for one minute (55°C for exons 15E and 15I), 72°C for one minute, and finally 72°C for 10 minutes. In cases of poor amplification, flanking primers for exon 15 were used to perform a nested PCR prior to using specific primers for the region of interest.
Single strand conformational polymorphism (SSCP) analysis
SSCP was performed as previously described.24 Electrophoresis was performed in 10% non-denaturing polyacrylamide gels and the PCR products visualised with silver staining using standard methods.
Direct sequencing of the MCR of the APC gene
The nucleotide sequences of the PCR products showing an abnormal electrophoretic mobility on SSCP analysis were determined by direct sequencing of purified PCR product in a thermocycle sequencing reaction with the dRhodamine Sequencing kit on a 377 prism sequencer (ABI). The sequences obtained from our experiments were performed in duplicate and alongside samples with known wild-type genotypes.
Immunohistochemistry
Immunohistochemistry was performed on sections from formalin fixed paraffin wax embedded tumour tissue using the labelled streptavidin method. Fresh 4 μm thick sections were dewaxed in xylene and rehydrated in graded alcohols. Endogenous peroxidase activity was blocked by 15 minutes of incubation in 3% hydrogen peroxide in distilled water. Antigen retrieval was achieved by microwaving in 0.01 M citrate buffer, pH 6, and the sections were incubated with mouse monoclonal antibodies under the conditions shown in table 2 ▶. A second layer of biotinylated rabbit antimouse antibody (1:300; Dako, Ely, Cambridge, UK) was applied for one hour followed by a final layer of horseradish peroxidase labelled streptavidin (1:500; Dako) for one hour. The bound antibody was detected using diaminobenzidine tetrahydrochloride as the chromogen. Negative controls (performed using phosphate buffered saline instead of primary antibody) and positive controls (consisting of tissue known to be positive for the relevant antibody) were included in every experiment.
Table 2.
Duration of microwaving for antigen retrieval, and concentration of primary antibody with duration of incubation
| Monoclonal antibody | Microwave duration (min) | Concentration of primary antibody, duration of incubation |
| hMLH1 | 30 | 1:100 (Pharmingen), 18 h |
| hMSH2 | 30 | 1:100 (Pharmingen), 18 h |
| β-catenin | 10 | 1:100 (Affiniti), 1 h |
| E-cadherin | 20 | Supernatant (ICRF), 1 h |
| p53 | 10 | 1:1000 (ICRF), 1 h |
Immunostaining of hMLH1 and hMSH2 in tumour cells was evaluated as present or absent. β-Catenin membrane staining was scored34 in accordance with the proportion of positive tumour cells as follows: less than 10%, homogenous 10–90% (with altered distribution—that is, cytoplasmic or nuclear), heterogenous 10–90%, and more than 90%. E-cadherin membranous staining was scored semiquantatively in accordance with the proportion of positive tumour cells as follows: 0%, 1–25%, 26–50%, and more than 50%. For p53 immunostaining, tumours were scored semiquantatively in accordance with the proportion of positive tumour cells as follows: less than 10%, 10–25%, 26–50%, and more than 50%.
RESULTS
RER status
One of 21 (5%) cancers was found to be RER+. This was a 58 year old male who underwent resection of a moderately differentiated T4 Nx adenocarcinoma of the ileum.
Screening for mutations in the APC gene
Despite screening of the whole MCR, no mutations were detected in the APC gene in this study. However, we detected two polymorphisms. These were silent changes at codon 1201 (TCA→TCG, Ser→Ser) and 1220 (ACA→ACG, Thr→Thr). Both polymorphisms were found in heterozygotes and have not been previously described.
Immunohistochemistry
hMLH1 and hMSH2
All cancers expressed hMLH1 and hMSH2 protein in the tumour cell nuclei (fig 1 ▶).
Figure 1.
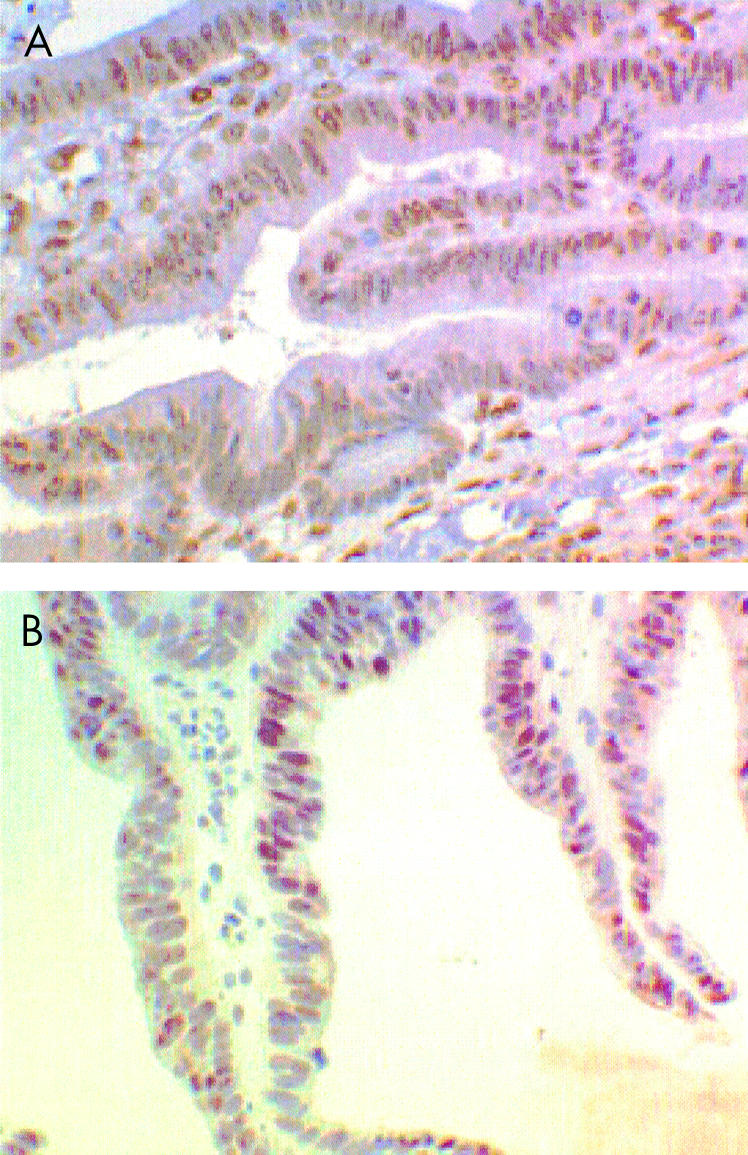
All cancers expressed hMLH1 (A) and hMSH2 (B) in the tumour cell nuclei. Only one of the 21 small bowel cancers was relication error positive, and this may have been due to a “gain of function” mutation in either hMLH1 and hMSH2, or a mutation in one of the other mismatch repair genes such as GTBP.
β-catenin
β-catenin staining was found along the intercellular borders of normal small bowel mucosa. Abnormal (reduced) expression of β-catenin was observed in 17 of 21 (81%) cancers (fig 2 ▶). Decreased membranous staining was seen in all 17 of these cancers, with increased cytoplasmic and nuclear staining observed in 14 (67%) and 10 (48%) cancers, respectively. Complete loss of membranous staining in association with nuclear staining was seen in only one cancer. Although all cancers with nuclear expression of β-catenin had reduced membrane staining, this was not statistically significant (Fisher's exact value, p=0.055).
Figure 2.
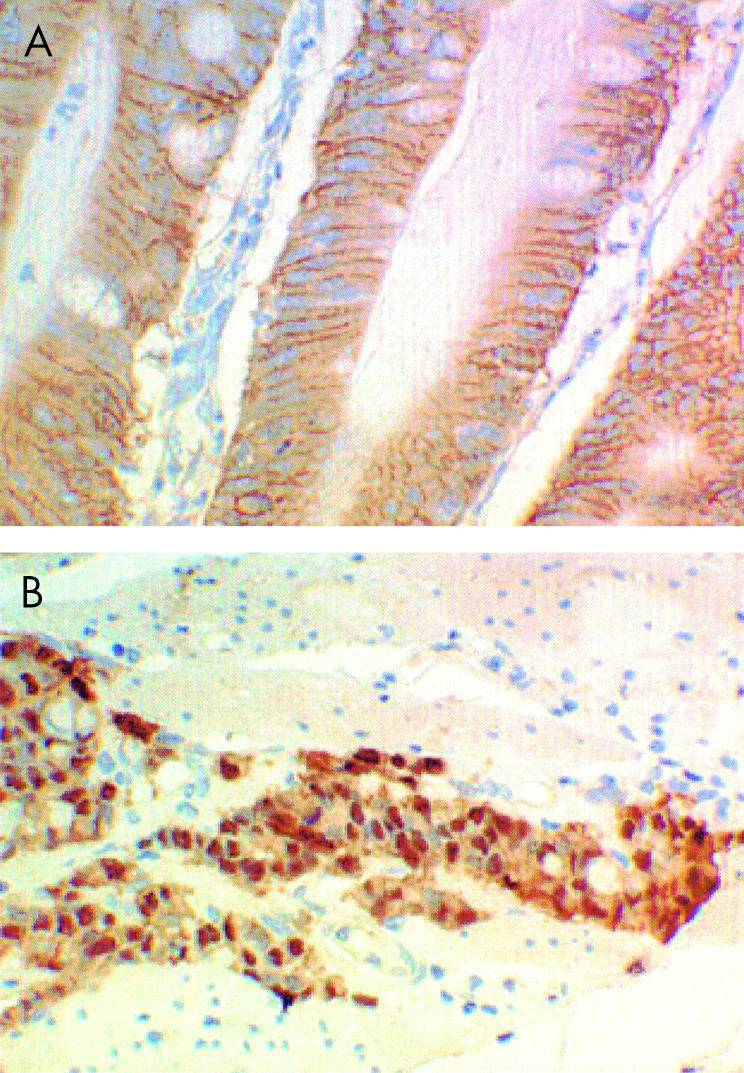
β-Catenin staining was found along the intercellular borders of normal small bowel mucosa and four cancers (A). Abnormal expression of β-catenin was observed in 17 of 21 (81%) cancers with decreased membranous staning and increased nuclear staining observed in 10 (48%) cancers (B).
E-cadherin
Uniform membranous staining of E-cadherin was localised at the intercellular borders in normal small bowel mucosa. Those cancers which showed membrane staining in over 25% of tumour cells were regarded as positive for E-cadherin expression. Reduced membrane expression of E-cadherin (<25% of tumour cells) was found in eight cancers (38%) (fig 3 ▶) with loss of cell surface staining in two of these cancers. Cytoplasmic staining, in the absence of membrane staining, was regarded as negative, as membrane localisation is essential for E-cadherin function. Although poorly differentiated cancers were more likely to have reduced membrane staining (four of six poorly differentiated cancers versus four of 15 moderately/well differentiated cancers), this was not statistically significant (Fisher's exact value, p=0.1).
Figure 3.
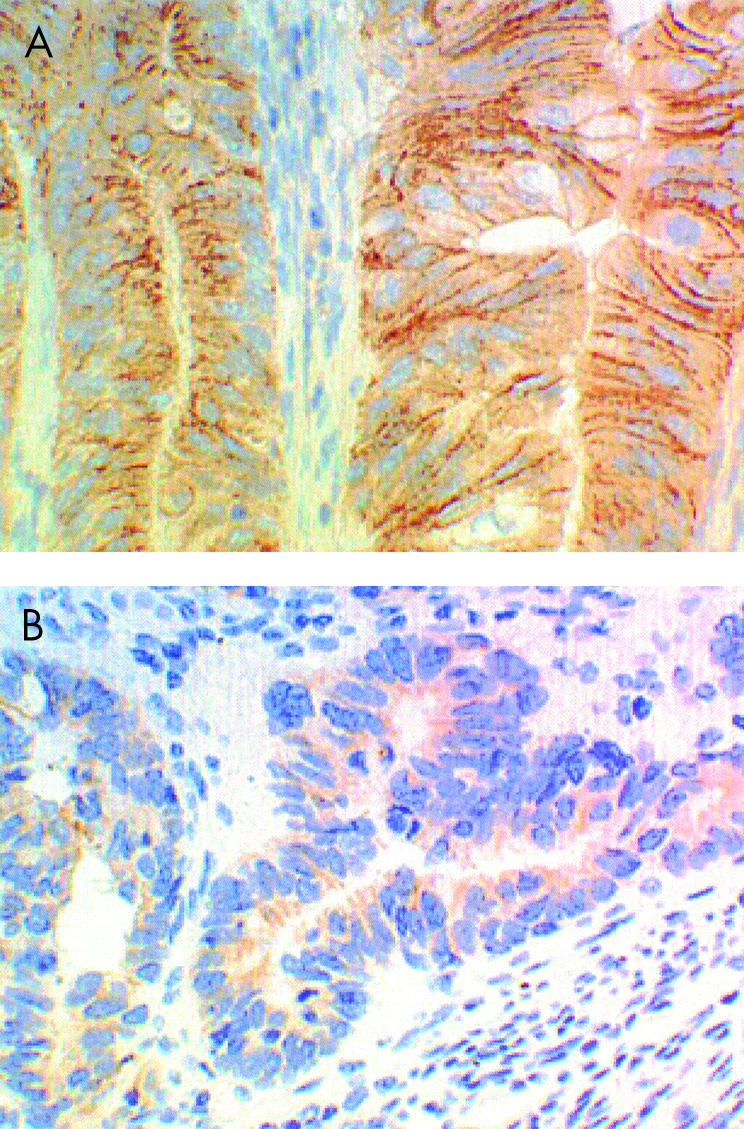
Uniform membranous staining of E-cadherin was found along the intercellular borders in normal small bowel mucosa and 13 cancers (A). Reduced membrane expression (<25% of tumour cells) was found in eight cancers (38%) (B).
p53
The p53 immunohistochemistry showed strong staining in the tumour cell nuclei. A cancer was regarded as positive for p53 overexpression if there was staining of over 10% of the tumour cells. Overexpression of p53 protein was detected in the nuclei of five of the 21 (24%) cancers (fig 4 ▶).
Figure 4.
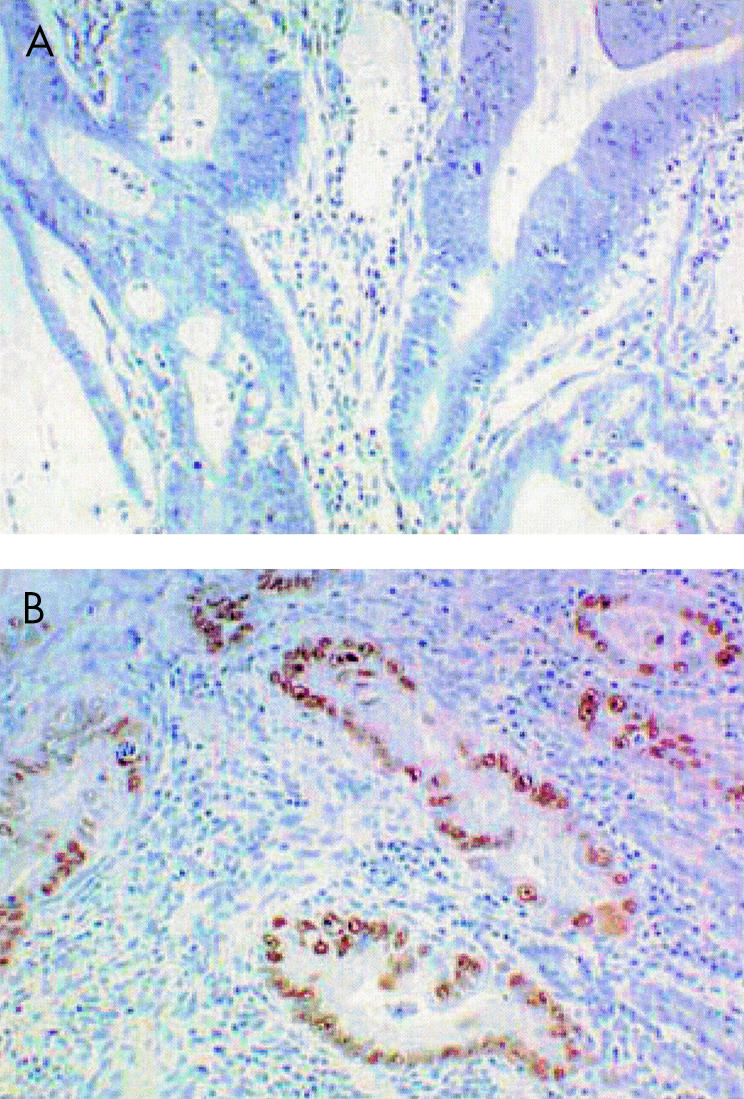
Wild type p53 exists at a level below the threshold of immunohistochemical detection (A). Overexpression of p53 protein was detected in the nuclei of five of 21 (24%) cancers (B).
DISCUSSION
We have screened a panel of 21 non-familial, non-ampullary small bowel adenocarcinomas for RER status, mutations in the APC MCR, and immunostaining of hMLH1, hMSH2, β-catenin, E-cadherin, and p53.
Only one (5%) of the cancers was found to be RER+. This is not dissimilar (allowing for the small sample size) to the 15% incidence of microsatellite instability in sporadic colorectal cancers or to the 13% incidence previously described in sporadic small bowel cancer.35 Previous work has reported microsatellite instability in five of 11 (45%) adenocarcinomas of the small intestine36 but as adenocarcinoma of the small intestine is 25 times more common in HNPCC kindreds,23 and patients with a positive family history of colorectal cancer were not excluded, selection bias may have accounted for this high value.
The one RER+ cancer stained positively for both hMLH1 and hMSH2 protein. This may be due to germline missense mutations which account for 33% of hMLH1 (and a smaller percentage of hMSH2) mutations found in HNPCC kindreds,37 although the patient did not have a family history suggestive of HNPCC. Alternatively, either a “gain of function” mutation may have occurred somatically or a mutation was present in one of the other MMR genes such as GTBP. Hypermethylation of the hMLH1 promoter region with loss of hMLH1 protein expression has recently been described as the principal cause of the RER+ phenotype in sporadic colorectal, endometrial, and gastric cancers with microsatellite instability.38–46 It would seem reasonable therefore to suggest that true RER+ sporadic adenocarcinomas of the small intestine may also sometimes have this epigenetic change.
We did not detect any mutations in the MCR of the APC gene. This is in keeping with Arai and colleagues47 who described only one nonsense mutation (an adenine insertion at codons 1554–1556, a common mutation in FAP patients with gastroduodenal cancers48) in a panel of 15 adenocarcinomas of the small intestine. Others have shown that allelic loss at 5q (the region of the APC gene) is rare in adenocarcinoma of the small intestine,35 and this is in contrast with the frequent allelic loss seen in colorectal cancers.49 It is possible that mutations would have been detected if we had screened the whole of the APC gene, but up to 80% of sporadic colorectal cancers have truncating mutations of APC,12 and 60% of these are within the MCR.24 There were two young patients in our series, aged only 24 and 21 years, and it is possible that these patients could have had a new mutation in the APC gene. As germline APC mutations may occur throughout the whole of the APC gene, it is possible that only screening the MCR missed a mutation in these two patients. Mutations in the APC gene have been described in other gastrointestinal malignancies (gastric and pancreatic cancer50–52), although these were not as common a finding as in colorectal cancer. Inactivation of APC may still be an early event in adenocarcinoma of the small intestine, and epigenetic changes, such as hypermethylation of the promoter region, may be responsible. Although hypermethylation of the APC promoter region has been studied briefly in colorectal cancer,53 this is an area of carcinogenesis that needs to be further investigated. The lack of a suitable antibody prevented us from studying expression of APC protein.
The β-catenin and E-cadherin genes are potential candidates (and an alternative to APC) that may cause derangement of cellular architecture in the early pathway of colorectal carcinogenesis.54 Ten (48%) cancers in this study had decreased membranous expression of β-catenin with a concomitant increase in nuclear staining. Although β-catenin is present in small amounts in the nucleus of normal colonic epithelium, this is below the level that is detectable by immunohistochemistry. The increased accumulation of β-catenin in 10 cancers reflects the prevention of β-catenin degradation and has been associated with the presence of mutant APC protein when it occurs in colorectal cancer.55 In the absence of APC mutations detected in this study, it is possible that the increased nuclear localisation could have been caused by β-catenin gain of function mutations which have been reported previously in up to 25% of colorectal cancers.27,28 β-catenin mutations have been described both in colorectal cancers with wild-type APC and in colorectal cancers with mutant APC. In the latter cancers, β-catenin mutations must augment the effect of the APC mutation in disrupting the control of the β-catenin signalling pathway,54 or else there is no basis for selection of the mutation.
Eight cancers (38%) in this series had decreased (<25% of tumour cells) membranous expression of E-cadherin and this is very similar to the incidence reported in colorectal cancers.56 As in colorectal and other cancers, this may be due to mutations or hypermethylation of the promoter in the E-cadherin gene.57–60 Although there was an association of poorly differentiated cancers with decreased membrane staining (as with colorectal cancers), this was not statistically significant.
Overexpression of p53 protein was detected in the nuclei of five of the 21 (24%) cancers. Arai et al detected p53 overexpression in eight of 15 (53.3%) adenocarcinomas of the small intestine,47 and in four of these cancers missense mutations were detected. Together with this previous study and findings in colorectal cancers, the results suggest that p53 mutation plays an important role in adenocarcinoma of the small intestine.
Other genes previously studied in adenocarcinoma of the small intestine include K-ras, where a similar incidence of point mutations has been reported to that found in colorectal cancer.2,47,61 However, Younes et al reported K-ras mutations in duodenal adenocarcinoma (four of 12 cancers) but did not find any mutations in adenocarcinoma of the jejunum and ileum (16 cancers).62
In conclusion, only one of our 21 (5%) adenocarcinomas of the small intestine was RER+. We did not detect mutations in the MCR of the APC gene, and this suggests that adenocarcinoma of the small intestine may follow a somewhat different genetic pathway to colorectal cancer, although still often involving alterations associated with β-catenin and E-cadherin. This is the first study to examine the role of β-catenin and E-cadherin in adenocarcinoma of the small intestine. Abnormal expression was common and may reflect an early pathway in which mutations may be found in adenocarcinoma of the small intestine. Overexpression of p53 is a relatively frequent finding and, as in colorectal cancer, reflects its important role in the carcinogenesis of the small intestine. These results give some insight into the genetic pathway of adenocarcinoma of the small intestine and show intriguing differences from colorectal cancers with respect to APC mutations, which may also be reflected in other cancers.
Abbreviations
FAP, familial adenomatous polyposis
HNPCC, hereditary non-polyposis colorectal cancer
APC gene, adenomatous polyposis coli gene
RER, replication error
MMR, mismatch repair
MCR, mutation cluster region
PCR, polymerase chain reaction
SSCP, single strand conformational polymorphism
REFERENCES
- 1.Gore RM. Small bowel cancer. Clinical and pathologic features. Radiol Clin North Am 1997;35:351–60. [PubMed] [Google Scholar]
- 2.Arber N, Neugut AI, Weinstein IB, et al. Molecular genetics of small bowel cancer. Cancer Epidemiol Biomarkers Prev 1997;6:745–8. [PubMed] [Google Scholar]
- 3.Ross RK, Hartnett NM, Bernstein L, et al. Epidemiology of adenocarcinomas of the small intestine: is bile a small bowel carcinogen? Br J Cancer 1991;63:143–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sellner F. Investigations on the significance of the adenoma-carcinoma sequence in the small bowel. Cancer 1990;66:702–15. [DOI] [PubMed] [Google Scholar]
- 5.Neugut AI, Santos J. The association between cancers of the small and large bowel. Cancer Epidemiol Biomarkers Prev 1993;2:551–3. [PubMed] [Google Scholar]
- 6.Neugut AI, Marvin MR, Rella VA, et al. An overview of adenocarcinoma of the small intestine. Oncology 1997;11:529–36. [PubMed] [Google Scholar]
- 7.Johnson AM, Harman PK, Hanks JB. Primary small bowel malignancies. Am Surg 1985;51:31–6. [PubMed] [Google Scholar]
- 8.Vasen HF, Mecklin JP, Khan PM, et al. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis Colon Rectum 1991;34:424–5. [DOI] [PubMed] [Google Scholar]
- 9.Potten CS, Li YQ, O'Connor PJ, et al. A possible explanation for the differential cancer incidence in the intestine, based on distribution of the cytotoxic effects of carcinogens in the murine large bowel. Carcinogenesis 1992;13:2305–12. [DOI] [PubMed] [Google Scholar]
- 10.Lowenfels AB. Why are small-bowel cancers so rare? Lancet 1973;1:24–26. [DOI] [PubMed] [Google Scholar]
- 11.Mittal VK, Bodzin JH. Primary malignant tumors of the small bowel. Am J Surg 1980;140:396–9. [DOI] [PubMed] [Google Scholar]
- 12.Bodmer W. The somatic evolution of cancer. The Harveian Oration of 1996. J R Coll Physicians Lond 1997;31:82–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell 1990;61:759–67. [DOI] [PubMed] [Google Scholar]
- 14.Peltomaki P. Microsatellite instability and hereditary non-polyposis colon cancer. J Pathol 1995;176:329–30. [DOI] [PubMed] [Google Scholar]
- 15.Akiyama Y, Sato H, Yamada T, et al. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res 1997;57:3920–3. [PubMed] [Google Scholar]
- 16.Miyaki M, Konishi M, Tanaka K, et al. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat Genet 1997;17:271–2. [DOI] [PubMed] [Google Scholar]
- 17.Fishel R, Lescoe MK, Rao MR, et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993;75:1027–38. [DOI] [PubMed] [Google Scholar]
- 18.Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993;75:1215–25. [DOI] [PubMed] [Google Scholar]
- 19.Bronner CE, Baker SM, Morrison PT, et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994;368:258–61. [DOI] [PubMed] [Google Scholar]
- 20.Papadopoulos N, Nicolaides NC, Wei YF, et al. Mutation of a mutL homolog in hereditary colon cancer. Science 1994;263:1625–9. [DOI] [PubMed] [Google Scholar]
- 21.Nicolaides NC, Papadopoulos N, Liu B, et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994;371:75–80. [DOI] [PubMed] [Google Scholar]
- 22.Papadopoulos N, Nicolaides NC, Liu B, et al. Mutations of GTBP in genetically unstable cells. Science 1995;268:1915–17. [DOI] [PubMed] [Google Scholar]
- 23.Watson P, Lynch HT. Extracolonic cancer in hereditary nonpolyposis colorectal cancer. Cancer 1993;71:677–85. [DOI] [PubMed] [Google Scholar]
- 24.Nagase H, Nakamura Y. Mutations of the APC (adenomatous polyposis coli) gene. Hum Mutat 1993;2:425–34. [DOI] [PubMed] [Google Scholar]
- 25.Munemitsu S, Albert I, Souza B, et al. Regulation of intracellullar B-catenin levels by the adenomatous polyposis coli (APC) tumour-suppressor protein. Proc Natl Acad Sci USA 1995;92:3046–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubinfield B, Albert I, Porfiri E, et al. Loss of B-catenin regulation by the APC tumour suppressor protein correlates with loss of structure due to common mutations of the gene. Cancer Res 1997;57:4624–30. [PubMed] [Google Scholar]
- 27.Morin PJ, Sparks AB, Korinek V, et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 1997;275:1787–90. [DOI] [PubMed] [Google Scholar]
- 28.Ilyas M, Tomlinson IP, Rowan A, et al. Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA 1997;94:10330–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ilyas M, Tomlinson IP. The interactions of APC, E-cadherin and beta-catenin in tumour development and progression. J Pathol 1997;182:128–37. [DOI] [PubMed] [Google Scholar]
- 30.Lane DP. p53 and human cancers. Br Med Bull 1994;50:582–9. [DOI] [PubMed] [Google Scholar]
- 31.Ilyas M, Straub J, Tomlinson IP, et al. Genetic pathways in colorectal and other cancers. Eur J Cancer 1999;35:335–51. [DOI] [PubMed] [Google Scholar]
- 32.Zhou XP, Hoang JM, Li YJ, et al. Determination of the replication error phenotype in human tumors without the requirement for matching normal DNA by analysis of mononucleotide repeat microsatellites. Genes Chromosomes Cancer 1998;21:101–7. [DOI] [PubMed] [Google Scholar]
- 33.Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell 1991;66:589–600. [DOI] [PubMed] [Google Scholar]
- 34.Valizadeh A, Karayiannakis AJ, El-Hariry I, et al. Expression of E-cadherin-associated molecules (a-, B-, and g-catenins and p120) in colorectal polyps. Am J Pathol 1997;150:1977–84. [PMC free article] [PubMed] [Google Scholar]
- 35.Rashid A, Hamilton SR. Genetic alterations in sporadic and Crohn's-associated adenocarcinomas of the small intestine. Gastroenterology 1997;113:127–35. [DOI] [PubMed] [Google Scholar]
- 36.Hibi K, Kondo K, Akiyama S, et al. Frequent genetic instability in small intestinal carcinomas. Jpn J Cancer Res 1995;86:357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peltomaki P, Vasen HF. Mutations predisposing to hereditary nonpolyposis colorectal cancer: database and results of a collaborative study. The International Collaborative Group on Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 1997;113:1146–58. [DOI] [PubMed] [Google Scholar]
- 38.Kane MF, Loda M, Gaida GM, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res 1997;57:808–11. [PubMed] [Google Scholar]
- 39.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA 1998;95:6870–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Veigl ML, Kasturi L, Olechnowicz J, et al. Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci USA 1998;95:8698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cunningham JM, Christensen ER, Tester DJ, et al. Hypermethylation of the hMLH1 promoter in colon cancer with microsatellite instability. Cancer Res 1998;58:3455–60. [PubMed] [Google Scholar]
- 42.Wheeler JMD, Beck NE, Kim HC, et al. Mechanisms of inactivation of mismatch repair genes in human colorectal cancer cell lines: the predominant role of hMLH1. Proc Natl Acad Sci USA 1999;96:10296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gurin CC, Federici MG, Kang L, et al. Causes and consequences of microsatellite instability in endometrial carcinoma. Cancer Res 1999;59:462–6. [PubMed] [Google Scholar]
- 44.Leung SY, Yuen ST, Chung LP, et al. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high-frequency microsatellite instability. Cancer Res 1999;59:159–64. [PubMed] [Google Scholar]
- 45.Fleisher AS, Esteller M, Wang S, et al. Hypermethylation of the hMLH1 gene promoter in human gastric cancers with microsatellite instability. Cancer Res 1999;59:1090–5. [PubMed] [Google Scholar]
- 46.Simpkins SB, Bocker T, Swisher EM, et al. MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum Mol Genet 1999;8:661–6. [DOI] [PubMed] [Google Scholar]
- 47.Arai M, Shimizu S, Imai Y, et al. Mutations of the Ki-ras, p53 and APC genes in adenocarcinomas of the human small intestine. Int J Cancer 1997;70:390–5. [DOI] [PubMed] [Google Scholar]
- 48.Toyooka M, Konishi M, Kikuchi-Yanoshita R, et al. Somatic mutations of the adenomatous polyposis coli gene in gastroduodenal tumors from patients with familial adenomatous polyposis. Cancer Res 1995;55:3165–70. [PubMed] [Google Scholar]
- 49.Powell SM, Zilz N, Beazer-Barclay Y, et al. APC mutations occur early during colorectal tumorigenesis. Nature 1992;359:235–7. [DOI] [PubMed] [Google Scholar]
- 50.Horii A, Nakatsuru S, Miyoshi Y, et al. Frequent somatic mutations of the APC gene in human pancreatic cancer. Cancer Res 1992;52:6696–8. [PubMed] [Google Scholar]
- 51.Horii A, Nakatsuru S, Miyoshi Y, et al. The APC gene, responsible for familial adenomatous polyposis, is mutated in human gastric cancer. Cancer Res 1992;52:3231–3. [PubMed] [Google Scholar]
- 52.Nakatsuru S, Yanagisawa A, Ichii S, et al. Somatic mutation of the APC gene in gastric cancer: frequent mutations in very well differentiated adenocarcinoma and signet-ring cell carcinoma. Hum Mol Genet 1992;1:559–63. [DOI] [PubMed] [Google Scholar]
- 53.Hiltunen MO, Alhonen L, Koistinaho J, et al. Hypermethylation of the APC (adenomatous polyposis coli) gene promoter region in human colorectal carcinoma. Int J Cancer 1997;70:644–8. [DOI] [PubMed] [Google Scholar]
- 54.Bodmer W. Familial adenomatous polyposis (FAP) and its gene, APC. Cytogenet Cell Genet 1999;86:99–104. [DOI] [PubMed] [Google Scholar]
- 55.Inomata M, Hayashi M, Ohno-Iwashita Y, et al. Involvement of calpain in integrin-mediated signal transduction. Arch Biochem Biophys 1996;328:129–34. [DOI] [PubMed] [Google Scholar]
- 56.Ilyas M, Tomlinson IP, Hanby A, et al. Allele loss, replication errors and loss of expression of E-cadherin in colorectal cancers. Gut 1997;40:654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Efstathiou JA, Liu D, Wheeler JMD, et al. Mutated epithelial cadherin is associated with increased tumorigenicity and loss of adhesion and of responsiveness to the motogenic trefoil factor 2 in colon carcinoma cells. Proc Natl Acad Sci USA 1999;96:2316–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wheeler JMD, Kim HC, Efstanthiou LA, et al. Hypermethylation of the promoter region of the E-cadherin gene (CDHI) in sporadic and ulcerative colitis associated colorectal cancer. Gut 2001;48:367–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berx G, Becker KF, Hofler H, et al. Mutations of the human E-cadherin (CDH1) gene. Hum Mut 1998;12:226–37. [DOI] [PubMed] [Google Scholar]
- 60.Graff JR, Herman JG, Lapidus RG, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res 1995;55:5195–9. [PubMed] [Google Scholar]
- 61.Sutter T, Arber N, Moss SF, et al. Frequent K-ras mutations in small bowel adenocarcinomas. Dig Dis Sci 1996;41:115–18. [DOI] [PubMed] [Google Scholar]
- 62.Younes N, Fulton N, Tanaka R, et al. The presence of K-12 ras mutations in duodenal adenocarcinomas and the absence of ras mutations in other small bowel adenocarcinomas and carcinoid tumors. Cancer 1997;79:1804–8. [PubMed] [Google Scholar]