

Abstract
Background: Tauroursodeoxycholate (TUDC) provides partial protection against taurolithocholate (TLC) induced cholestasis, possibly by inducing a signalling cascade activating protein kinase C (PKC). The potential protective effects of β muricholic acid (β-MC), another 7-β-hydroxylated bile salt, have not previously been studied in TLC cholestasis.
Aims: To study the effect of β-MC on TLC induced cholestasis and also to investigate further the effects of agents affecting intracellular signalling, notably DBcAMP (a cell permeable cAMP analogue) and several protein kinase inhibitors.
Methods: Functional studies were carried out analysing the proportion of hepatocyte couplets able to accumulate the fluorescent bile acid analogue cholyl-lysyl-fluorescein (CLF) into their sealed canalicular vacuole (cVA of CLF assay).
Results: It was found that both β-MC and DBcAMP were as effective as TUDC in protecting against TLC induced cholestasis. The PKC inhibitors staurosporin and H7 but not the specific protein kinase A (PKA) inhibitor KT5720 abolished the protective effects of TUDC and β-MC. BAPTA/AM, a chelator of intracellular Ca2+, significantly decreased the protective effect of both bile salts, and that of DBcAMP. PKC and PKA inhibitors had no effect on protection with DBcAMP.
Conclusions: β-MC was as effective as TUDC in protecting against TLC cholestasis. Mobilisation of Ca2+ and activation of PKC, but not of PKA, are involved in the anticholestatic effect of the two 7-β-hydroxylated bile salts. The hepatoprotective effects of DBcAMP involved Ca2+ mobilisation, but not PKC or PKA activation.
Keywords: cholestasis, ursodeoxycholate, muricholate, DBcAMP cell signalling, hepatocyte couplets
Ursodeoxycholic (UDC) and its taurine (TUDC) or glycine conjugates demonstrate hepatoprotective properties,1 both experimentally and in the treatment of patients with cholestatic disorders.2 UDC and its conjugates protect against liver cell damage induced by high doses of more hydrophobic bile salts—for example, taurocholate,3 chenodeoxycholate,4–6 and deoxycholate.6 TUDC also counteracted reduction of bile flow in experimental models of drug induced cholestasis, including oestrogens7 their glucuronoconjugates,7, 8 cyclosporin A,9 and taurolithocholate (TLC).10
The mechanism(s) underlying the beneficial anticholestatic effects of UDC is still debated. This bile salt and its conjugates have been shown to have membrane stabilising effects protecting against bile salt induced membrane solubilisation.11 UDC amide conjugates stimulate bile salt excretion and, as accumulation of bile salts may play a crucial role in cholestasis associated liver damage, improvement of bile salt extrusion may attenuate cholestasis.2, 3 Finally, UDC has been shown to protect against bile salt induced mitochondrial dysfunction,12, 13 oxidative stress,13, 14 and apoptosis,14, 15 three causally associated features in cholestasis.16
It is increasingly evident that UDC and its conjugates are signalling molecules with distinctive regulatory properties on signal transduction pathways. UDC, when present in low concentrations, is unique among the bile salts in evoking long lasting intracellular Ca2+ oscillations in hepatocytes by stimulating both Ca2+ release from microsomal inositol-1,4,5-trisphosphate (IP3) sensitive stores via an IP3 independent pathway, and Ca2+ influx from extracellular sources via Ni2+ sensitive channels.17–19 TUDC also selectively stimulated vesicular transport by the transcytotic pathway, indicated by the sustained increase in horseradish peroxidase (HRP) excretion into bile.18
Stimulation of this or other vesicular pathways via Ca2+ elevation may lead to increased targeting and insertion of transport proteins into the canalicular membrane.20 In line with this we have recently shown that permeable adenosine 3`:5`-cyclic monophosphate (cAMP) and N6,2`-o-dibutyryladenosine 3`:5`-cyclic monophosphate (dibutyryl cAMP or DBcAMP), stimulated targeting of canalicular transporters by a Ca2+ dependent mechanism.21 Beuers et al have suggested that TUDC may activate an intracellular signalling cascade leading to activation of the calcium dependent protein kinase C (PKC) isoform, PKC-α.2 PKC activation increased vesicular fusion and exocytosis,18, 22, 23 claimed to stimulate targeting and insertion of canalicular carriers into their membrane domain.19 Finally, although UDC does not directly affect cAMP levels or protein kinase A (PKA) activity in hepatocytes, it shares with cAMP a number of hepatoprotective properties, including the ability to protect against hydrophobic bile salt induced cytolytic damage,5 apoptosis,24 or oxidative stress.25 Like UDC,17 cAMP increases cytosolic Ca2+ levels in hepatocytes26, 27 and induces sustained excretion of HRP into bile.28 Furthermore, DBcAMP stimulates membrane domain targeting and transport activity of the canalicular transporters: multidrug resistance protein 2 (mrp2, a non-bile salt organic anion transporter),21, 29, 30 mdr2 (a “flippase” translocating phospholipids),29 mrp1 (an organic cation transporter),29 and the Cl−/HCO3− exchanger.31 Bile salt transport activity, assessed using a fluorescent bile salt analogue in hepatocyte couplets, was also stimulated by DbcAMP.21, 32
Despite the striking similarities between DBcAMP and UDC as signalling molecules and their abilities to stimulate biliary secretion and target canalicular transporter delivery to specific membrane domains, the role of DBcAMP as an anticholestatic agent, in addition to its well recognised effect as a anticytotoxic compound (see above), has never been investigated and represents one of the aims of the present study.
For this purpose, TLC was used as a model cholestatic compound. Monohydroxylated bile salts such as TLC, although present only as trace amounts in normal bile, are thought to play an important role in cholestatic diseases in which their hepatic levels are enhanced—for example, in severe neonatal cholestasis,33 parenteral nutrition induced cholestasis,34 and chenodeoxycholate therapy.35
Like UDC, tauro-β-muricholate is a bile acid possessing a 7-β-hydroxyl, and has been shown to preserve choleresis and to prevent hydrophobic bile acid induced hepatotoxicity and cholestasis both in normal rats5, 36 and in rats treated with the microtubule disrupter colchicine.37 Another major aim of this study was therefore to investigate whether β muricholate (β-MC), in common with TUDC, can prevent TLC induced cholestasis, and whether these mechanisms of hepatoprotection involve PKC, PKA, and Ca2+ dependent signal transduction cascades.
METHODS
Materials
Cholyl-lysyl-fluorescein (CLF) was synthesised according to Mills and colleagues.38, 39 Collagenase (type A) was purchased from Gibco (Paisley, Scotland) and β-MC from Steraloids Inc (Newport, USA). Bovine serum albumin was purchased from Winlab (Maidenhead, UK), TUDC, TLC, staurosporine (SP), 1,2-bis-(o-aminophenoxy)-ethene-N,N,N`,N`-tetra-acetate tetra-(acetomethyl)ester (BAPTA/AM), H7, DBcAMP, KT2057, Leibovitz-15 tissue culture medium (L-15), and other fine chemicals from Sigma (Poole, UK).
Animals
Male Wistar rats (230–240 g body weight) had free access to a standard laboratory diet (41B maintenance diet; Pilsbury, Birmingham UK) and tap water ad libitum. Surgery was performed between 8 and 9 am to minimise circadian variations. Anaesthesia was achieved with ketamine hydrochloride 6 mg/100 g body weight and medetomidine 25 μg/100 g body weight. Protocols were approved according to the Animals Scientific Procedures Act 1986.
Isolation, enrichment, and culture of hepatocytes
Hepatocyte couplets were obtained from rat liver according to Wilton and colleagues,40 adapted from Gautam and colleagues.41 The final preparation had a viability >90% (trypan blue exclusion) and contained 25–35% couplets. It was enriched using centrifugal elutriation to 70–80% couplets, with viability >95%. Couplets were plated in L-15 medium containing penicillin/streptomycin in 35 mm plastic culture dishes (2 ml per dish) at 0.5×106 units/ml, and incubated at 37°C for 4.5 hours prior to experimentation.
Assessment of canalicular vacuolar accumulation (cVA)
Couplets were incubated for 15 minutes at 37°C with 2 μM CLF (final concentration). The fluorescent bile salt was then rapidly washed away twice with 2 ml L-15 medium and couplets were immediately examined using an inverted fluorescence microscope (IMT2-RFL; Olympus, London, UK). The number of couplets in four randomly selected regions of the plate (150–250 in each experiment) accumulating CLF into a fluorescent canalicular vacuole, judged by visual inspection as a proportion of the total couplets analysed, was then determined. This parameter is referred to as “cVA of CLF”, and is an indication of overall biliary secretion—that is, uptake, transcellular transport, canalicular secretion, and intracanalicular retention. Examples of a fully functional couplet preparation (control), a TLC inhibited preparation, and a preparation protected from TLC by TUDCA are shown in fig 1 ▶. The couplets counted as positive are those showing enough accumulation to be visualised (thresholded by the eye); within this population there may be a range of different accumulating abilities representing different original positions in the acinus, different expression of transporters, etc. Change in the proportion of accumulating couplets in the population represents a shift in the range of the couplet abilities. This semiquantitative approach enables rapid examination of the secretory behaviour of a mixed population of hepatocyte couplets and therefore is more likely to reflect what happens to the whole liver than analysis of the performance of an individual couplet. Furthermore, it provides similar information to other more quantitative but time consuming alternatives, using image analysis. Indeed, quantitative estimation of the amount of CLF retained in the canalicular vacuole (estimated as the product between mean canalicular fluorescence intensity and canalicular area corrected by total couplet fluorescence) showed a close correlation with semiquantitative estimation of the proportion of canalicular vacuoles displaying a visible amount of CLF.42
Figure 1.
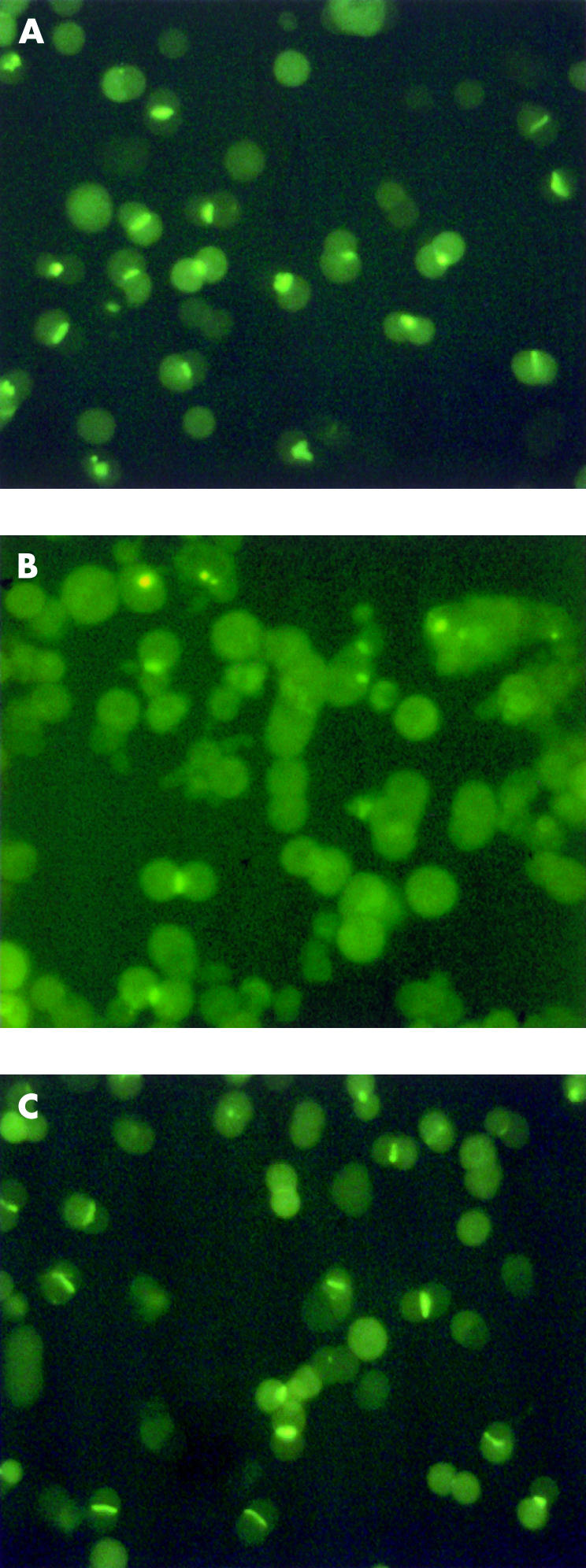
Hepatocyte couplets accumulating cholyl-lysyl-fluorescein (CLF) in the technique of canalicular vacuolar accumulation (cVA) of CLF. (A) Couplet preparation after 15 minutes of incubation with CLF. (B) Couplets exposed to 2.5 μM taurolithocholate (TLC); subsequently incubated with CLF for 15 minutes. (C) Couplets coincubated with TLC and tauroursodeoxycholate (TUDCA) (2 μM); subsequently incubated with CLF for 15 minutes.
Estimation of the influence of BAPTA/AM, H7, SP, and KT5720 on cVA of CLF
Couplets were incubated for 30 minutes with 10 μl of dimethyl sulphoxide (DMSO) (vehicle control), 20 μM BAPTA/AM (a Ca2+ chelator) (concentrations represent final concentrations in the incubation medium), 5 μM KT5720 (a specific PKA inhibitor), 100 μM H7 (a PKC/PKA inhibitor), and 1 μM SP (a pan-specific PKC inhibitor), as appropriate. CVA of CLF was analysed as described above. Concentrations of these compounds were chosen by making a balance between their effectiveness and specificity, based on Km and Ki inhibitory concentrations for their respective substrates, as described by the manufacturers.
Estimation of the influence of inhibitors on the cholestatic effect of TLC
Couplets were preincubated with 10 μl of DMSO (vehicle control), 20 μM BAPTA/AM, 100 μM H7, 1 μM SP, or 5 μM KT5720 (as appropriate) for 15 minutes. Then, TLC (2.5 μM) was added, and cells were incubated for another 30 minutes; cVA of CLF was then analysed as described above.
Estimation of the effect of TUDC, β-MC, and DBcAMP on TLC induced cholestasis
Couplets were incubated for 30 minutes with 10 μl of DMSO (vehicle control), TLC (2.5 μM), TLC (2.5 μM)+β-MC (2 μM), TLC (2.5 μM)+TUDC (2 μM), or TLC (2.5 μM)+DBcAMP (10 μM), as appropriate. All concentrations were derived from preliminary studies (data not shown). CLF (2 μM) was then added and incubated at 37°C for 15 minutes before washing twice with 2 ml of L-15; couplets accumulating CLF were counted as described above.
Estimation of the effect of inhibitors on hepatoprotection with TUDC and β-MC in TLC induced cholestasis
Couplets were preincubated with 10 μl of DMSO (vehicle control), 20 μM BAPTA/AM, 100 μM H7, 1 μM SP, or 5 μM KT5720 for 15 minutes. Then, TLC (2.5 μM), TLC (2.5 μM)+TUDC (2 μM), or TLC (2.5 μM)+β-MC (2 μM), as appropriate, was added, and cells were incubated for another 30 minutes. cVA of CLF was then analysed as described above. A schematic representation depicting the mechanism by which TUDC, DBcAMP, and the different signal modulators used in this study would exert modulatory effects on both signal transduction pathways and recycling of canalicular transporters from the subapical compartment (SAC) to the canalicular membrane, is shown in fig 2 ▶.
Figure 2.

Schematic representation of the mechanisms of regulation of hepatocellular signal transduction by the hepatoprotective agents (TUDC, β-MC, and DBcAMP). TUDC, and presumably β-MC, increase cytosolic Ca2+ levels by stimulating both Ca2+ release from microsomal inositol-1,4,5-trisphosphate (IP3) sensitive stores via an IP3 independent pathway and Ca2+ influx from extracellular sources via Ni2+ sensitive channels,17–19 leading to activation of the Ca2+ dependent PKC isoform, PKC-α.2 DBcAMP, a permeable cAMP analogue, is instrumental in both elevating cytosolic Ca2+ and activating PKA. Activation of Ca2+, PKC, and PKA dependent pathways may mediate reinsertion of canalicular transporters by stimulating their recycling from the SAC; this may counteract localisation changes of these carriers induced by TLC. To assess the role of these different signal transduction pathways, several inhibitors have been used, including the intracellular Ca2+ chelator BAPTA/AM, the PKC inhibitors H7 and SP, and the PKA inhibitor KT5720. BAPTA/AM, 1,2-bis-(o-aminophenoxy)-ethene-N,N,N`,N`-tetra-acetate tetra-(acetomethyl)ester; DBcAMP, dibutyryl-cAMP; β-MC, β-muricholate; PKC-α, protein kinase C α; PKA, protein kinase A; SAC, subapical compartment; SP, staurosporine; TUDC, tauroursodeoxycholate.
Estimation of the effects of inhibitors on hepatoprotection with DBcAMP in TLC induced cholestasis
Couplets were preincubated with 10 μl of DMSO (vehicle control), 20 μM BAPTA/AM, 1 μM SP, 5 μM KT5720, or BAPTA/AM (20 μM)+KT5720 (5 μM) for 15 minutes. Then, TLC (2.5 μM) alone, or together with DBcAMP (10 μM), was added, and cells were incubated for another 30 minutes. cVA of CLF was then analysed as described above.
Statistical analysis
Results are expressed as mean (SD). Statistical comparisons were made using ANOVA. Differences were considered significant when p<0.05.
RESULTS
TLC induced a significant decrease in cVA of CLF (from 57 (3)% to 25 (7)%, p<0.001). Significant protection from this impairment was given by TUDC (increase from cVA of 25 (7)% to 41 (9)%; p<0.001), β-MC (increase from cVA of 25 (4)% to 42 (5)%; p<0.001), and DBcAMP (increase from cVA of 25 (7)% to 41 (13)%; p<0.001). These represented relative increases in comparison with the unprotected TLC induced level of 64%, 68%, and 64% for TUDC, β-MC, and DBcAMP, respectively.
When both TUDC and DBcAMP were used together, their effect was not greater than for each compound used alone (from cVA of 25 (7)% to 36 (11)%; p<0.01); a 44% increase compared with controls (table 1 ▶).
Table 1.
Effect of TUDC, β-MC, and DBcAMP on the cholestasis induced by TLC
| Control | TLC | TLC+TUDC | TLC+β-MC | TLC+DBcAMP | TLC+TUDC+DBcAMP | |
| cVA of CLF (%) | 57 (3) | 25 (7) | 41 (9)*** | 42 (5)*** | 41 (13)*** | 36 (11)** |
Hepatocyte couplets were incubated for 30 minutes at 37°C with DMSO (controls), TLC, TLC+TUDC, TLC+βMC, TLC+DBcAMP, and TLC+TUDC+DBcAMP. cVA of CLF was then analysed, as described in the methods section.
All values are mean (SD) (n=5–6).
TUDC, β-MC, and DBcAMP had a significant protective effect against TLC induced cholestasis. No additive protective effect was observed when both TUDC and DBcAMP where administered together.
Significantly different from TLC, **p<0.01, ***p<0.001.
CLF, cholyl-lysyl-fluorescein; DBcAMP (dibutyryl cAMP), N6,2`-o-dibutyryladenosine 3`:5`-cyclic monophosphate; DMSO, dimethyl sulphoxide; cVA, canalicular vacuolar accumulation; β-MC, β-muricholate; TLC, taurolithocholate; TUDC, tauroursodeoxycholate.
The protective effect of TUDC was significantly impaired by the presence of the Ca2+ chelator BAPTA/AM, the PKC/PKA inhibitor H7, and the PKC inhibitor SP. The specific PKA inhibitor KT5720 had no significant effect however on hepatoprotection with TUDC (fig 3 ▶).
Figure 3.

Effect of inhibitors of protein kinase C (PKC), protein kinase A (PKA), and Ca2+ dependent signalling pathways on hepatoprotection induced by tauroursodeoxycholate (TU) against taurolithocholate (TLC) induced decrease in canalicular vacuolar accumulation (cVA) of cholyl-lysyl-fluorescein (CLF). Couplets were incubated for 15 minutes at 37°C with dimethyl sulphoxide (DMSO, control), 1,2-bis-(o-aminophenoxy)-ethene-N,N,N`,N`-tetra-acetate tetra-(acetomethyl)ester (BAPTA/AM (B)), H7, staurosporine (S), or KT5720 (KT). Then, TLC was added to dishes containing DMSO, and TLC+TU were added to the remaining dishes, and the cells were incubated for another 30 minutes. cVA of CLF was then analysed, as described in the methods section. Values are means (SD) (n=5–6). The protective effect of TU on TLC induced cholestasis was significantly decreased by the presence of B, H7, or S, but not by KT. Significantly different from TLC, ***p<0.001; significantly different from TLC+TU, ††p<0.01, †††p<0.001; NS, not significant.
In the absence of the hepatoprotective compounds, PKC and PKA inhibitors did not have any significant independent effect on cVA of CLF (57 (3)%, 56 (7)%, 55 (3)%, and 58 (10)% for BAPTA/AM, H7, SP, and KT5720, respectively, compared with 61 (7)% in controls; NS). Similarly, in the absence of the hepatoprotective compounds, these signalling modulators did not influence the ability of TLC to impair cVA of CLF in hepatocyte couplets (25 (7)%, 24 (14)%, 22 (5)%, 28 (3)%, and 22 (6)% for TLC+BAPTA/AM, TLC+H7, TLC+SP, and TLC+KT5720, respectively, compared with 25 (7)% in TLC alone; p<0.05) (tables 2, 3 ▶ ▶).
Table 2.
Effect of the inhibitors of PKC, PKA, and Ca2+ dependent signalling pathways used in this study on cVA of CLF
| Control | BAPTA/AM | H7 | SP | KT5720 | |
| cVA of CLF (%) | 61 (7) | 57 (3) | 56 (7) | 55 (3) | 58 (10) |
Hepatocyte couplets were incubated for 30 minutes at 37°C with DMSO (control), BAPTA/AM, H7, SP, or KT5720, before adding CLF and assessing cVA of CLF, as described in the methods section.
All values are mean (SD); n= 4–6 separated set of couplet preparations.
Differences between groups were not statistically significant.
CLF, cholyl-lysyl-fluorescein; BAPTA/AM, 1,2-bis-(o-aminophenoxy) -ethene-N,N,N`,N`-tetra-acetate tetra-(acetomethyl)ester; DMSO, dimethyl sulphoxide; cVA, canalicular vacuolar accumulation; PKA, protein kinase A; PKC, protein kinase C; SP, staurosporine.
Table 3.
Effect of the inhibitors of PKC, PKA, and Ca2+dependent signalling pathways used in this study on TLC induced decrease in cVA of CLF
| TLC | TLC+ BAPTA/AM | TLC+H7 | TLC+SP | TLC+KT5720 | |
| cVA of CLF (%) | 25 (7) | 24 (14) | 22 (5) | 28 (3) | 22 (6.3 |
Couplets were incubated for 15 minutes at 37°C with DMSO (control), BAPTA/AM, H7, SP, or KT5720. Then, TLC was added to the dishes containing DMSO, and TLC was added to the remaining dishes, and cells were incubated for another 30 minutes. cVA of CLF was then analysed, as described in the methods section.
All values are mean (SD); n= 5–6 separated set of couplet preparations.
Differences between groups were not statistically significant.
CLF, cholyl-lysyl-fluorescein; BAPTA/AM, 1,2-bis-(o-aminophenoxy) -ethene-N,N,N`,N`-tetra-acetate tetra-(acetomethyl)ester; DMSO, dimethyl sulphoxide; cVA, canalicular vacuolar accumulation; PKA, protein kinase A; PKC, protein kinase C; SP, staurosporine; TLC, taurolithocholate.
The protective effect of β-MC was also reduced, although to a lesser extent, by the Ca2+ chelator BAPTA/AM, the PKC/PKA inhibitor H7, and the PKC inhibitor SP. Again, the PKA inhibitor KT5720 did not affect the improvement in cVA of CLF caused by β-MC (fig 4 ▶).
Figure 4.

Effect of inhibitors of protein kinase C (PKC), protein kinase A (PKA), and Ca2+ dependent signalling pathways on hepatoprotection induced by β-muricholate (MA) against taurolithocholate (TLC) induced decrease in canalicular vacuolar accumulation (cVA) of cholyl-lysyl-fluorescein (CLF). Couplets were incubated for 15 minutes at 37°C with dimethyl sulphoxide (DMSO, control), 1,2-bis-(o-aminophenoxy)-ethene-N,N,N`,N`-tetra-acetate tetra-(acetomethyl)ester (BAPTA/AM (B)), H7, staurosporine (S), or KT5720 (KT). Then, TLC was added to dishes containing DMSO, and TLC+MA were added to the remaining dishes, and cells were incubated for another 30 minutes. cVA of CLF was then analysed. All values are means (SD) (n=5–6). The protective effect of MA on TLC induced cholestasis was significantly decreased by the presence of B, H7, and S, but not by KT. Significantly different from TLC, ***p<0.001; significantly different from TLC+MA, †p<0.05; NS, not significant.
BAPTA/AM, but not SP or KT5720, significantly impaired the protective effect of DBcAMP. When both BAPTA/AM and KT5720 were administered together, their effects were not different from those of BAPTA alone (fig 5 ▶).
Figure 5.

Effect of inhibitors of protein kinase C (PKC), protein kinase A (PKA), and Ca2+ dependent signalling pathways on hepatoprotection induced by dibutyryl cAMP (cAMP) against taurolithocholate (TLC) induced decrease in canalicular vacuolar accumulation (cVA) of cholyl-lysyl-fluorescein (CLF). Couplets were incubated for 15 minutes at 37°C with dimethyl sulphoxide (DMSO, control), 1,2-bis-(o-aminophenoxy)-ethene-N,N,N`,N`-tetra-acetate tetra-(acetomethyl)ester (BAPTA/AM (B)), staurosporine (S), KT5720 (KT), or B+KT. Then, TLC was added to the dishes containing DMSO, and TLC+DBcAMP were added to the remaining dishes, and cells were incubated for another 30 minutes. cVA of CLF was then analysed. All values are means (SD) (n=5–6). The protective effect of DBcAMP on TLC induced cholestasis was significantly decreased by the presence of B but not by S or KT. When both KT and B were added together, their inhibiting effect was not greater than B alone. Significantly different from TLC, ***p<0.001; NS, not significant; significantly different from TLC+cAMP, †p<0.05.
DISCUSSION
Stimulation of hepatocellular secretion by UDC and its conjugates via selective activation of different signalling pathways has been proposed to play a key role in their anticholestatic effects,2, 20, 22, 43, 44 but the hypothesis has yet to be tested in experimental models of cholestasis. In this study, we examined the putative mediation of three signal transduction cascades—that is, PKA, PKC, and Ca2+ dependent signalling pathways—in the hepatoprotective effects of two bile salts, TUDC and β-MC, against TLC induced cholestasis.
The study revealed:
TUDC was not unique in preventing TLC induced cholestasis as β-MC, another 7β-hydroxylated bile salt, was as effective as TUDC in exhibiting anticholestatic properties;
BAPTA/AM, a chelator of intracellular Ca2+, significantly decreased the protective effect of both bile salts, supporting the contention that the mechanisms by which they exert their hepatoprotective effects involve Ca2+ mobilisation;
TUDC and β-MC induced improvement of fluorescent bile salt accumulation was abolished by the PKC inhibitors SP and H7, but not by the specific PKA inhibitor KT5720, suggesting that activation of PKC is involved in the anticholestatic effect of both 7β-hydroxylated bile salts;
DBcAMP also attenuated TLC induced impairment of cVA of CLF, but its beneficial effect was only counteracted by the Ca2+ chelator BAPTA/AM and not by the PKA inhibitor KT5720, or the PKC inhibitor SP, indicating that mobilisation of Ca2+, but not PKA or PKC activation, is involved in this hepatoprotective action of DBcAMP.
TLC induces cholestasis by inhibiting both bile salt dependent and independent fractions of bile flow.45 The mechanism by which TLC impairs bile salt secretion remains unclear. An alteration in the bile salt export pump, the canalicular transporter involved in bile salt secretion into the canalicular space, is likely as this represents the limiting step in overall hepatic bile salts transport.12 Roma et al have shown that TLC induced cholestasis selectively impairs canalicular transfer of the mrp2 substrate sulphobromophthalein both in vivo and in isolated hepatocytes, without affecting its uptake.46
Thus TUDC may stimulate vesicle mediated insertion of canalicular carriers waiting to be delivered into their membrane domain, and thus counteract reversal of this process induced by TLC exposure. Our finding that TUDC exerts its anticholestatic effect by Ca2+ dependent PKC activation fits well with this contention. Indeed, PKC activation with the phorbol ester phorbol dibutyrate stimulates exocytosis of previously accumulated HRP, a marker of transcytotic vesicular delivery, in isolated perfused liver.23 Also, a similar stimulation of exocytic activity occurs by translocation/subsequent activation of PKC from cytosol to membranes in rat pancreatic β cells.47, 48 A subcellular fraction enriched in microtubule associated vesicles containing both canalicular export pumps and the transcytotic marker polymeric immunoglobulin A receptor, has been recently characterised; this vesicular pool may represent, or be functionally related to, a subapical vesicle pool involved in delivery of canalicular carriers.49 Roelofsen et al showed that phorbol ester induced PKC activation favours recycling (following endocytosis induced by hepatocyte isolation) of canalicular transporters back to the plasma membrane in isolated hepatocytes50 This is in line with the finding that PKC stimulates efflux of the mrp2 substrate dinitrophenyl-S-glutathione,51 and the organic cation tri-n-buthylmethylamonium,52 in isolated hepatocytes. Furthermore, phorbol ester stimulated PKC-α translocation, insertion of vesicles into the Golgi apparatus, and increased vesicular transport occurs in HepG2 cells.53 In contrast, inhibition by PKC of spontaneous and DbcAMP stimulated transcytotic targeting of canalicular carriers was observed, both following restoration of couplet polarity post-isolation21 and in DbcAMP stimulated targeting of the Cl− /HCO3− exchanger to the canalicular membrane in isolated liver cells.31 Taken together, these results suggest that distinct pathways exist for vesicular trafficking of transporters to the canalicular region which are differentially regulated by signal modulators, with the one stimulated by PKC being activated by TUDCA and β-MC. Whether this anticholestatic action is mediated by PKC activation itself or by other effectors of the PKC activated signalling cascade downstream of PKC remains to be answered. The involvement of PKC mediated activation of mitogen activated protein kinases (MAPK), suggested to mediate TUDC induced stimulation of bile salt secretion, is unlikely as simultaneous inhibition of PKC did not block TUDC induced MAPK activation54; this suggests that other factors able to activate MAPKs, such as cytosolic Ca2+,55 are involved.
Our results show that Ca2+ dependency for TUDC and β-MC induced hepatoprotection was indirect, via activation of Ca2+ dependent PKC isoforms, presumably PKC-α, activated by TUDC.22 This conclusion arises from the present studies showing that the anticholestatic effects of the hepatoprotective bile salts were fully counteracted by the two PKC inhibitors as well as by the Ca2+ chelator BAPTA/AM (see fig 2 ▶). However, our finding that DBcAMP exerted its effects exclusively via Ca2+ elevation—that is, in a PKA and PKC independent way (see fig 5 ▶), support the contention that, in addition to PKC activation, Ca2+ elevation per se affords hepatoprotection against TLC induced cholestasis. These apparently discrepant results are not necessarily in contradiction as under conditions of PKC activation (as occurs following TUDC and, presumably, β-MC, administration), many DBcAMP stimulated apically directed vesicular transport processes have been reported to be inhibited, including: increased targeting of canalicular transporters into their canalicular domain21, 31; HRP biliary excretion28; and sorting of sphingolipids to the apical pole.56 Ca2+ mediated hepatoprotection may therefore not be operating when PKC is simultaneously activated by administration of TUDC or β-MC. Alternatively, Ca2+ mediated hepatoprotection may require the coadjuvant effect of other signalling pathways activated by DBcAMP, such as PKA activation, to induce its beneficial effect.
In conclusion, our results show that both TUDC and β-MC can protect disruption of biliary secretory function in hepatocyte couplets. PKC activation seems to play a crucial role in this beneficial effect. DBcAMP, which shares with TUDC some properties as a signal molecule, also exhibits a similar anticholestatic effect. This effect occurs via Ca2+ elevation but, in contradistinction, is independent of PKC activation, which is crucial to TUDC and β-MC afforded hepatoprotection in this model. Further studies of the mechanisms underlying the anticholestatic effect of this family of hepatoprotective bile salts should provide an insight into the events that, subsequent to Ca2+ elevation and/or PKC activation, mediate their hepatoprotective functions.
Acknowledgments
We would like to thank Sir Jules Thorne Charitable Trust (PM), Sir Samuel Scott of Yews Trust (PM), Wellcome Trust (MR), and the Royal Society (MR) for financial support during various aspects of this work.
Abbreviations
cAMP, adenosine 3`:5`-cyclic monophosphate
CLF, cholyl-lysyl-fluorescein
DBcAMP (dibutyryl cAMP), N62`-o-dibutyryladenosine 3`:5`-cyclic monophosphate
BAPTA/AM, 1,2-bis-(o-aminophenoxy)-ethene-N,N,N`,N,-tetra-acetate tetra-(acetomethyl)ester
DMSO, dimethyl sulphoxide
cVA, canalicular vacuolar accumulation
HRP, horseradish peroxidase
mrp, multidrug resistance protein
PKA, protein kinase A
IP3, inositol-1,4,5-trisphosphate
L-15, Leibovitz-15
MAPKs, mitogen activated protein kinases
β-MC, β muricholate
PDB, phorbol 12,13-dibutyrate
PKC, protein kinase C
SP, staurosporine
SAC, subapical compartment
TLC, taurolithocholate
TUDC, tauroursodeoxycholate
UDC, ursodeoxycholate
Footnotes
Presented in part at the annual 51st AASLD Meetings, Dallas, Texas, USA, November 2000; published in abstract form (Hepatology 2000;32(pt 2):429A).
REFERENCES
- 1.Kitani K. The protective effect of hydrophilic bile acids on bile acid hepatotoxicity in the rat. Ital J Gastroenterol 1995;27:366–71. [PubMed] [Google Scholar]
- 2.Beuers U, Beuer JL, Paumgartenr G. Ursodeoxycholic acid in cholestasis: potential mechanisms of action and therapeutic applications. Hepatology 1998;28:1449–53. [DOI] [PubMed] [Google Scholar]
- 3.Kitani K, Kanai S. Tauroursodeoxycholate prevents taurocholate induced cholestasis. Life Sci 1982;30:515–23. [DOI] [PubMed] [Google Scholar]
- 4.Kanai S, Ohta M, Kitani K, et al. Tauro β-muricholate is as effective as tauroursodeoxycholate in preventing taurochenodeoxycholate-induced liver damage in the rat. Life Sci 1990;47:2421–8. [DOI] [PubMed] [Google Scholar]
- 5.Ohiwa T, Katagiri K, Hoshino M, et al. Tauroursodeoxycholate and tauro-beta-muricholate exert cytoprotection by reducing intrahepatic taurochenodeoxycholate content. Hepatology 1993;17:470–6. [PubMed] [Google Scholar]
- 6.Heuman DM, Mills AS, McCall J, et al. Conjugates of ursodeoxycholate protect against cholestasis and hepatocellular necrosis caused by more hydrophobic bile acids. In vivo studies in the rat. Gastroenterology 1991;100:203–11. [DOI] [PubMed] [Google Scholar]
- 7.Bouchard G, Yousef IM, Tuchweber B. Influence of oral treatment with ursodeoxycholic and tauroursodeoxycholic acids on estrogen-induced cholestasis in rats: effects on bile formation and liver plasma membranes. Liver 1993;13:193–202. [DOI] [PubMed] [Google Scholar]
- 8.Kinbara S, Ishizaki K, Sakakura H, et al. Improvement of estradiol-17 beta-D-glucuronide-induced cholestasis by sodium tauroursodeoxycholate therapy in rats. Scand J Gastroenterol 1997;32:947–52. [DOI] [PubMed] [Google Scholar]
- 9.Queneau PE, Bertault Peres P, Guitaoui M, et al. Improvement of cyclosporin A-induced cholestasis by tauroursodeoxycholate in a long-term study in the rat. Dig Dis Sci 1994;39:1581–5. [DOI] [PubMed] [Google Scholar]
- 10.Schölmerich J, Baumgartner U, Miyai K, et al. Tauroursodeoxycholate prevents taurolithocholate-induced cholestasis and toxicity in rat liver. J Hepatol 1990;10:280–3. [DOI] [PubMed] [Google Scholar]
- 11.Heuman DM, Bajaj R. Ursodeoxycholate conjugates protect against disruption of cholesterol-rich membranes by bile acids. Gastroenterology 1994;106:1333–41. [DOI] [PubMed] [Google Scholar]
- 12.Krähenbühl S, Fischer S, Talos C, et al. Ursodeoxycholate protects oxidative mitochondrial metabolism from bile acid toxicity: dose-response study in isolated rat liver mitochondria. Hepatology 1994; 20:1595–601. [DOI] [PubMed] [Google Scholar]
- 13.Rodrigues CM, Fan G, Wong PY, et al. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol Med 1998;4:165–78. [PMC free article] [PubMed] [Google Scholar]
- 14.Rodrigues CM, Fan G, Ma X, et al. A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J Clin Invest 1998;101:2790–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Benz C, Angermüller S, Otto G, et al. Effect of tauroursodeoxycholic acid on bile acid-induced apoptosis in primary human hepatocytes. Eur J Clin Invest 2000;30:203–9. [DOI] [PubMed] [Google Scholar]
- 16.Rodrigues CM, Steer CJ. Mitochondrial membrane perturbations in cholestasis. J Hepatol 2000;32:135–41. [DOI] [PubMed] [Google Scholar]
- 17.Beuers U, Nathanson MH, Boyer JL. Effect of tauroursodeoxycholic acid on cytosolic Ca2+ signals in isolated rat hepatocytes. Gastroenterology 1993;104:604–12. [DOI] [PubMed] [Google Scholar]
- 18.Beuers U, Nathanson MH, Isales CM, et al. Tauroursodeoxycholic acid stimulates hepatocellular exocytosis and mobilizes extracellular Ca++, mechanisms defective in cholestasis. J Clin Invest 1993;92:2984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bouscarel B, Fromm H, Nussbaum R. Ursodeoxycholate mobilizes intracellular Ca2+ and activates phosphorylase a in isolated hepatocytes. Am J Physiol 1993;264:G243–51. [DOI] [PubMed] [Google Scholar]
- 20.Beuers U. Effects of bile acids on hepatocellular signaling and secretion. Yale J Biol Med 1997;70:341–6. [PMC free article] [PubMed] [Google Scholar]
- 21.Roma MG, Milkiewicz P, Elias E, et al. Control by signaling modulators of the sorting of canalicular transporters in rat hepatocyte couplets: Role of the cytoskeleton. Hepatology 2000;32:1342–56. [DOI] [PubMed] [Google Scholar]
- 22.Beuers U, Throckmorton DC, Anderson MS, et al. Tauroursodeoxycholic acid activates protein kinase C in isolated rat hepatocytes. Gastroenterology 1996;110:1553–63. [DOI] [PubMed] [Google Scholar]
- 23.Bruck R, Nathanson MH, Roelofsen H, et al. Effects of protein kinase C and cytosolic Ca2+ on exocytosis in the isolated perfused rat liver. Hepatology 1994;20:1032–40. [DOI] [PubMed] [Google Scholar]
- 24.Webster CR, Anwer MS. Cyclic adenosine monophosphate-mediated protection against bile acid-induced apoptosis in cultured rat hepatocytes. Hepatology 1998;27:1324–31. [DOI] [PubMed] [Google Scholar]
- 25.Nakano H, Monden M, Umeshita K, et al. Cytoprotective effect of prostaglandin I2 analogues on superoxide-induced hepatocyte injury. Surgery 1994;116:883–9. [PubMed] [Google Scholar]
- 26.Exton JH. Role of phosphoinositides in the regulation of liver function. Hepatology 1988;8:152–66. [DOI] [PubMed] [Google Scholar]
- 27.Staddon JM, Hansford RG. 4 β-Phorbol 12-myristate 13-acetate attenuates the glucagon-induced increase in cytoplasmic free Ca2+ concentration in isolated rat hepatocytes. Biochem J 1986;238:737–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayakawa T, Bruck R, Ng OC, et al. DBcAMP stimulates vesicle transport and HRP excretion in isolated perfused rat liver. Am J Physiol 1990;259:G727–35. [DOI] [PubMed] [Google Scholar]
- 29.Gatmaitan ZC, Nies AT, Arias IM. Regulation and translocation of ATP-dependent apical membrane proteins in rat liver. Am J Physiol 1997;272:G1041–9. [DOI] [PubMed] [Google Scholar]
- 30.Roelofsen H, Soroka CJ, Keppler D, et al. Cyclic AMP stimulates sorting of the canalicular organic anion transporter (Mrp2/cMoat) to the apical domain in hepatocyte couplets. J Cell Sci 1998;111:1137–45. [DOI] [PubMed] [Google Scholar]
- 31.Benedetti A, Strazzabosco M, Ng OC, et al. Regulation activity and apical targeting of the Cl-/HCO3- exchanger in rat hepatocytes. Proc Natl Acad Sci USA 1994;91:792–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyer Jl, Soroka CJ. Vesicle targeting to the apical domain regulates excretory function in isolated rat hepatocyte couplets. Gastroenterology 1995;109:1600–11. [DOI] [PubMed] [Google Scholar]
- 33.Setchell KD, Schwarz M, OConnell NC, et al. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7α-hydroxylase gene causes severe neonatal liver disease. J Clin Invest 1998;102:1690–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fouin Fortunet H, Le Quernec L, Erlinger S, et al. Hepatic alterations during total parenteral nutrition in patients with inflammatory bowel disease: a possible consequence of lithocholate toxicity. Gastroenterology 1982; 82:932–7. [PubMed] [Google Scholar]
- 35.Phillips MJ, Fisher RL, Anderson DW, et al. Ultrastructural evidence of intrahepatic cholestasis before and after chenodeoxycholic acid therapy in patients with cholelithiasis: the national cooperative gallstone study. Hepatology 1983;3:209–20. [PubMed] [Google Scholar]
- 36.Kanai S, Ohta M, Kitani K, et al. Tauro β-muricholate is as effective as tauroursodeoxycholate in preventing taurochenodeoxycholate-induced liver damage in the rat. Life Sci 1990; 47:2421–8. [DOI] [PubMed] [Google Scholar]
- 37.Katagiri K, Nakai T, Hoshino M, et al. Tauto-beta-muricholate preserves choleresis and prevents taurocholate induced cholestasis in colchicine treated rats. Gastroenterology 1992;102:1660–7. [DOI] [PubMed] [Google Scholar]
- 38.Mills CO, Rahman, Coleman R, et al. Cholyl-lysylfluorescein: synthesis, biliary excretion in vivo and during single-pass perfusion of isolated perfused rat liver. Biochim Biophys Acta 1991;1115:151–6. [DOI] [PubMed] [Google Scholar]
- 39.Mills CO, Milkiewicz P, Saraswat V, et al. Cholyllysyl fluorescein and related lysylfluorescein conjugated bile acid analogues. Yale J Biol Med 1997;70;447–57 [PMC free article] [PubMed] [Google Scholar]
- 40.Wilton JC, Williams DE, Strain AJ, et al. Purification of hepatocyte couplets by centrifugal elutriation. Hepatology 1991;14:180–3. [DOI] [PubMed] [Google Scholar]
- 41.Gautam A, Ng OC, Boyer JL. Isolated rat hepatocyte couplets in short term culture: structural characteristics and plasma membrane reorganisation. Hepatology 1987;7:216–23. [DOI] [PubMed] [Google Scholar]
- 42.Roma MG, Orsler D, Coleman R. Canalicular retention as a marker of tight junctional permeability in isolated hepatocyte couplets: Effects of protein kinase modulation and cholestatic agents. Fundam Appl Toxicol 1997;37:71–81. [DOI] [PubMed] [Google Scholar]
- 43.Bouscarel B, Kroll SD, Fromm H. Signal transduction and hepatocellular bile acid transport: cross talk between bile acids and second messengers. Gastroenterology 1999;117:433–52. [DOI] [PubMed] [Google Scholar]
- 44.Müller M, Jansen PLM. Molecular aspects of hepatobiliary transport. Am J Physiol 1997;272:G1285–303. [DOI] [PubMed] [Google Scholar]
- 45.Javitt NB, Emerman S. Effect of sodium taurolithocholate on bile flow and bile acid excretion. J Clin Invest 1968;47:1002–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roma MG, Penalva GL, Agüero RM, et al. Hepatic transport of organic anions in taurolithocholate-induced cholestasis in rats. J Hepatol 1994;20:603–10. [DOI] [PubMed] [Google Scholar]
- 47.Ganesan S, Calle R, Zawalich K, et al. Glucose induced translocation of protein kinase C in rat pancreatic islets. Proc Natl Acad Sci USA 1990;87: 9893–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ganesan S, Calle R, Zawalich K, et al. Immunocytochemical localisation of alpha protein kinase C in rat pancreatic beta cells during glucose induced insulin secretion. J Cell Biol 1992;119:313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Soroka CJ, Pate MK, Boyer JL. Canalicular export pumps traffic with polymeric immunoglobulin A receptor on the same microtubule-associated vesicle in rat liver. J Biol Chem 1999;274:26416–24. [DOI] [PubMed] [Google Scholar]
- 50.Roelofsen H, Bakker CTM, Schoemaker B, et al. Redistribution of canalicular organic anion transport activity in isolated and cultured rat hepatocytes. Hepatology 1995;21:1649–57. [PubMed] [Google Scholar]
- 51.Roelofsen H, Ottenhoff R, Oude Elferink RP, et al. Hepatocanalicular organic-anion transport is regulated by protein kinase C. Biochem J 1991;278:637–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steen H, Smit H, Nijholt A, et al. Modulators of the protein kinase C system influence biliary excretion of cationic drugs. Hepatology 1993;18:1208–15. [PubMed] [Google Scholar]
- 53.Westermann P, Knoblich M, Maier O, et al. Protein kinase C bound to the Golgi apparatus supports the formation of constitutive transport vesicles. Biochem J 1996;320:651–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schliess F, Kurz AK, vom Dahl S, et al. Mitogen-activated protein kinases mediate the stimulation of bile acid secretion by tauroursodeoxycholate in rat liver. Gastroenterology 1997;113:1306–14. [DOI] [PubMed] [Google Scholar]
- 55.Romanelli A, van de Werve G. Activation of mitogen-activated protein kinase in freshly isolated rat hepatocytes by both a calcium- and a protein kinase C-dependent pathway. Metabolism 1997;46:548–55. [DOI] [PubMed] [Google Scholar]
- 56.Zegers MM, Zaal KJ, van IJzendoorn SC, et al. Actin filaments and microtubules are involved in different membrane traffic pathways that transport sphingolipids to the apical surface of polarized HepG2 cells. Mol Biol Cell 1998;9:1939–49. [DOI] [PMC free article] [PubMed] [Google Scholar]