

Abstract
Background and aims: Mucosal flattening and epithelial cell apoptosis are typical features of T cell induced inflammatory diseases of the bowel, such as coeliac disease and graft versus host disease. Mice injected with a T cell activating anti-CD3 antibody develop a severe diarrhoeal illness. We describe the histological features of this enteropathy and define the effector mechanisms involved in T cell induced mucosal injury in this in vivo model.
Methods: Wild-type and genetically modified mice were injected with the anti-CD3 antibody 3C11 (50 μg). Changes in the murine intestine were characterised by light microscopy analysis and terminal uridine nick-end labelling (TUNEL) assay. The role of perforin, Fas/Fas ligand (FasL), tumour necrosis factor α (TNF-α), and interferon γ (IFN-γ) in T cell induced mucosal damage was assessed using selected immunodeficient mouse strains.
Results: T cell activation caused severe damage, including small intestinal mucosal flattening and apoptosis of crypt epithelial cells. Mucosal damage was unaltered in anti-CD3 treated mice lacking IFN-γ, Fas, or TNF-α receptors. In mice lacking TNF-α receptors and Fas (TNF-R1×R2 lpr/lpr strain), enterocyte apoptosis was diminished but there was no significant reduction in tissue damage. Apoptosis and mucosal injury were significantly reduced in perforin knockout mice. Abrogation of both FasL and perforin (perforin KO×gld mice) further significantly reduced tissue damage and apoptotic bodies.
Conclusions: T cell induced mucosal injury is mediated by the combined effect of multiple pathways but predominantly by perforin. The redundancy of the mechanisms of tissue damage will have significant impact on therapeutic strategies aimed at specific and targeted inhibition of inflammatory processes.
Keywords: enteropathy, T cell, tumour necrosis factor, Fas, perforin, mucosal damage, mouse intestine
In inflammatory diseases of the intestine (for example, coeliac disease and intestinal graft versus host (GvH) disease), the architectural changes of the mucosa have been attributed to local T cell activation.1–4 The architectural changes associated with mucosal damage, particularly epithelial cell apoptosis, villus flattening, and crypt hyperplasia can be reproduced experimentally by T cell activation in an in vitro model using human fetal intestinal explants,1 or in in vivo mouse models of GvH disease.5–7 The mechanisms by which T cell activation results in tissue damage and cell death of enterocytes in vivo have not been clarified to date. Circumstantial evidence suggests that release of tumour necrosis factor α (TNF-α),8 interferon γ (IFN-γ),9–11 and Fas ligand (FasL)3,12–15 are involved in mediating mucosal damage in T cell dependent intestinal diseases. Activation of local enzymes such as matrix metalloproteinases16,17 may also contribute.
In this study, we describe an in vivo mouse model of T cell mediated small intestinal tissue damage that is characterised by enterocyte apoptosis, epithelial damage, and villus atrophy. We assessed the molecular mechanisms responsible in vivo for T cell induced epithelial cell damage. We specifically investigated the individual and combined roles of the cytotoxic mediators TNF-α, IFN-γ, perforin, and FasL.
MATERIALS AND METHODS
Mice
Balb/c, C57BL/6, and MPJ mice (6–16 weeks of age), IFN-γ−/− mice, and Fas deficient MPJ lpr/lpr mice were obtained from the Jackson Laboratory (Bar Harbour, Maine, USA). SCID.bg mice were kindly provided by A Croy (University of Guelph); TNF-α receptor p55/TNF-R1−/− mice by Dr T Mak (Amgen Institute, Toronto, Canada); TNF-α receptor p75/TNF-R2-/- mice, mice deficient in both TNF-R1 and R2, and TNF-R1×R2 lpr/lpr mice by Dr J Peschon (Immunex Corporation, Seattle, Washington, USA). Generation of perforin KO/gld mice has been described previously.18 All mice were housed under specific pathogen free conditions. This study was approved by the McMaster University Animal Care Committee and conformed to the guidelines of the Canadian Council on Animal Care.
Monoclonal antibodies
Antimouse CD3ɛ antibody (Ab) (145–2C11) was provided by Dr J Bluestone (University of Chicago, Chicago, Illinois, USA). This Ab induces initial T cell activation and cytokine release followed by T cell inactivation and elimination and resulting immunosuppression.19 Hamster IgG (HIgG) control Ab was purchased from Rockland Inc. (Gilbertsville, Pennsylvania, USA).
Evaluation of in vivo anti-CD3 treatment
Mice received a single intraperitoneal injection of 50 μg of anti-CD3 or control Ab diluted in 500 μl of phosphate buffered saline (PBS), pH 7.4. In wild-type mice, this treatment reliably induced diarrhoea without being lethal. In some experiments, cyclosporin A (CsA 50 mg/kg) or dexamethasone (Dex 50 mg/kg) was given intraperitoneally either as a single dose at the same time as the anti-CD3 Ab or daily for a total of three injections beginning at the time of anti-CD3 injection. Mice were monitored for clinical signs and sacrificed at varying time points. Haematoxylin-eosin stained tissue sections of paraffin embedded intestinal specimens were graded in a blinded fashion using a quantitative histology score based on the frequency of apoptotic epithelial cells within the epithelium and the ratio of villus height to crypt length (table 1 ▶).
Table 1.
Small intestinal damage score
| Score | ||||
| 0 | 1 | 2 | 3 | |
| Villus height:crypt length ratio* | >3.5 | 2.5–3.5 | 1.5–2.4 | <1.5 |
| Apoptotic bodies† | 1–5 | 5–13 | 14–21 | >22 |
Damage score=sum of villus:crypt points and apoptosis points (maximal score=6).
*Mean of 5 villus/crypt units.
†Number of apoptotic bodies per 5 villus crypt units.
Apoptosis was assessed by identification of apoptotic bodies on histology and by identification of DNA fragmentation using terminal uridine nick-end labelling (TUNEL) staining on paraffin embedded sections. TUNEL staining was performed using the FragEL TDT detection kit (Calbiochem, Cambridge, Massachusetts, USA). Tissue samples from HIgG treated mice served as negative controls and irradiated thymocytes as positive controls.
Cytokine measurements
TNF-α and IFN-γ levels were measured by ELISA in duplicate serum samples (IFN-γ, Amersham International, Little Chalfont, Buckinghamshire, UK; TNF-α, R&D Systems, Minneapolis, MInnesota, USA). Tissue levels of TNF-α were measured in whole small intestine suspended in 5 ml of RPMI, homogenised, and then subjected to ultrasonication.
Detection of T cell activation
To assess mononuclear cell activation, cells were isolated 20 hours after anti-CD3 injection and six hours after bromodeoxyuridine (BrdU) injection (1.5 mg/mouse intraperitoneally). Intraepithelial lymphocytes (IEL) were isolated as previously described20 (viability of isolated IEL >95%), fixed in 70% ethanol at −20°C, permeabilised, and nuclear DNA denatured with 2 N HCl in 0.5% (v/v) Triton X-100 (Sigma, St Louis, Missouri, USA) solution. After neutralisation (0.1 M Na2B4O7, pH 8.5) and washing (0.5% Tween 20/1.0% bovine serum albumin/PBS, pH 7.4), samples were treated with either FITC conjugated anti-BrdU Ab (Becton Dickinson, San Jose, California, USA) or isotype control Ab, diluted 1:20 in Tween/bovine serum albumin/PBS. FITC fluorescence was measured on a FACScan (Becton Dickinson) using a doublet discrimination module set up to disregard cell aggregates.
Statistical analysis
Data are expressed as mean (SEM). Significance of differences in the quantitative intestinal damage score between groups was compared using the non-parametric Mann-Whitney U test as some data sets did not pass the Komolgorov-Smirnov test for normal distribution. Differing cytokine levels between groups were analysed using the Student's t test. Correlation of parameters was calculated by linear regression analysis using the SigmaStat computer program (Jandel Scientific Corp., Chicago, Illinois, USA).
RESULTS
Anti-CD3 induced enteropathy
Balb/c mice injected intraperitoneally with anti-CD3 Ab developed diarrhoea within four hours. At autopsy, 20 hours post injection, the small intestine was hyperaemic and contained liquid stool. Histological alterations of the small intestinal mucosa were characterised by reduced villus height, increased thickness of the crypt region, loss of Paneth cells, goblet cells, and IEL in the epithelial layer, and severe morphological changes of the epithelial cells (fig 1 ▶). In the villi, enterocytes lost their columnar and polarised morphology and became flattened. In the crypt region, numerous apoptotic bodies were identified in the epithelium. Regression analysis confirmed that reduction of villus:crypt ratio correlated closely with the number of apoptotic bodies in the epithelium (p=0.002). Apoptotic bodies disintegrated into small cell fragments that were cleared by infiltrating histiocytes within days. No inflammatory infiltrate of lymphocytic or granulocytic cells developed. By 72 hours post injection, the normal villus-crypt architecture eventually became an almost flat epithelium. A broadened layer of connective tissue replaced the apoptotic crypts. The intestinal damage score was maximal 20 hours after injection of anti-CD3 antibody (figs 1, 2 ▶ ▶). Villus-crypt architecture was almost completely restored by five days after anti-CD3 injection.
Figure 1.

Micrographs of the small intestinal mucosa from Balb/c mice treated with anti-CD3 or hamster IgG control antibody (40×). (A) Mucosa 20 hours after treatment with anti-CD3. The villi are shortened and oedematous; goblet cells and intraepithelial lymphocytes (IEL) are depleted. In the crypt regions, epithelial cells are difficult to distinguish and many apoptotic bodies can be found. Note villus height, short crypts, goblet cells, and IEL. (B) After 40 hours, villi are short and plump. Apoptotic bodies have disintegrated into nuclear fragments. (C) Mucosa 72 hours after anti-CD3 treatment. (D) Five days after anti-CD3 treatment, the mucosa appears normal. Terminal uridine nick-end labelling (TUNEL) assay demonstrates DNA fragmentation as a hallmark of apoptosis of epithelial cells 12 hours after anti-CD3 injection ((E) 40× and (F) 10× magnification).
Figure 2.

Time course of small intestinal mucosal damage in Balb/c mice treated with 50 μg of anti-CD3 antibody 145-2C11. (A) A significant increase (p<0.05) in damage score was observed beginning eight hours post injection (data are mean (SEM)). Tissue damage was highly significant (p<0.005) from 24 hours to three days post injection. Tissue damage had completely resolved by five days post injection. The damage score of mice treated with hamster IgG control antibody was below 1 for all time points (data not shown; n=3–6). (B) The number of apoptotic bodies increased significantly after anti-CD3 treatment (p<0.005 at 24 hours to three days post injection). (C) The ratio of villus height to crypt length was significantly reduced by 14 hours post injection (p<0.005 at 24 hours to three days post injection). *p<0.05 compared with controls; NS, no significant difference compared with controls (numbers indicate the number of animals treated).
In the large intestine, apoptosis of enterocytes was also detected but architectural changes remained minimal. Mucosal damage observed after anti-CD3 treatment of MPJ and C57BL/6 mice, the background strains for the knockout mice used in some of the subsequent experiments, was similar to that in Balb/c mice (fig 3 ▶).
Figure 3.

Damage score of wild-type background strains 24 hours after treatment with either hamster IgG (HIgG) control antibody (left) or anti-CD3 antibody (right). The histological damage that developed in the small intestine 24 hours after treatment with anti-CD3 antibody of C57Bl/6 mice and MPJ mice was similar to that observed in Balb/c mice (A). The number of apoptotic bodies was significantly higher in MPJ mice compared with both Balb/c or C57Bl/6 mice (B). *p<0.05 compared with MPJ mice treated with anti-CD3 antibody (numbers indicate the number of animals treated).
Demonstration of epithelial cell apoptosis
TUNEL staining showed that enterocyte cell death was preceded by DNA fragmentation (fig 1 ▶), suggesting apoptosis as the main mechanism of cell death and contributing to mucosal damage. TUNEL positive labelled epithelial cells were evident in the small intestinal (jejunal) crypts 12 hours after injection of anti-CD3 antibody. In the villi, TUNEL positive epithelial cells were not identified. No positive staining was observed in tissues taken from HIgG treated control animals.
Anti-CD3 induced enteropathy is accompanied by activation of local T cells
Local activation of T cells was demonstrated by increased DNA synthesis. IEL were isolated 20 hours after anti-CD3 treatment from mice that had also received an injection of the uridine analogue BrdU six hours prior to sacrifice. Incorporation of BrdU was detected by flow cytometry (fig 4 ▶).
Figure 4.
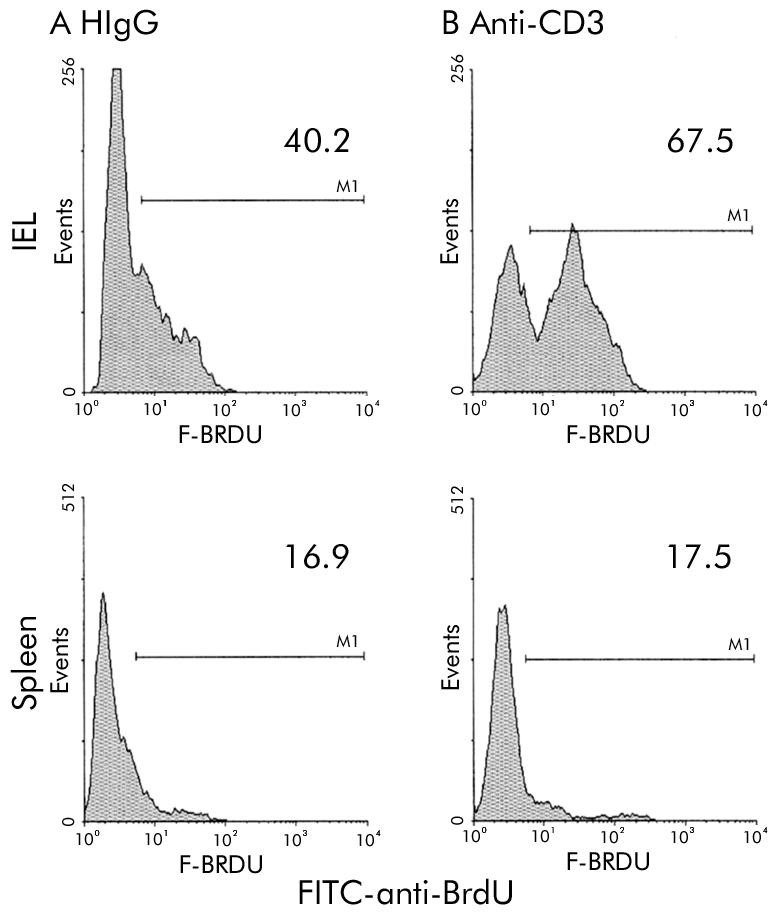
Activation of intraepithelial lymphocytes (IEL) in vivo is shown by demonstration of increased DNA synthesis (incorporation of uridine analogue bromodeoxyuridine (BrdU)). Single colour histograms are shown depicting the proportion of IEL that stained positive with FITC anti-BrdU antibody. (A) IEL isolated from mice treated with hamster IgG (HIgG) control antibody. (B) IEL isolated from mice 24 hours after treatment with anti-CD3 antibody.
Anti-CD3 induced enteropathy is prevented by immunosuppressive treatment
A single dose of 50 mg/kg CsA significantly reduced the anti-CD3 induced tissue damage observed 24 hours post injection. Similarly, Dex (50 mg/kg intraperitoneally), given at the time of anti-CD3 treatment, abrogated the development of severe mucosal damage (fig 5 ▶). Given daily for three days beginning at the time of administration of anti-CD3 antibody, immunosuppressive agents largely prevented tissue damage compared with mice treated solely with anti-CD3 antibody (fig 5 ▶). SCID mice lacking T cells21 failed to develop intestinal damage after treatment with anti-CD3 antibody (fig 7 ▶).
Figure 5.

Immunosuppressive treatment significantly reduced tissue damage induced by T cell activation. Treatment with a single dose of either cyclosporin A (CsA) or dexamethasone (Dex) significantly reduced the damage score observed 24 hours post injection (A). The damage score in mice treated with daily doses of either CsA or Dex was not different from control mice treated with only hamster IgG (HIgG) control antibody (B). *p<0.05 compared with control mice treated with only anti-CD3 antibody; †p<0.05 compared with control mice treated with only HIgG control antibody; NS, no significant difference compared with mice treated with HIgG control antibody. Damage score was assessed three days post injection (numbers indicate the number of animals treated).
Figure 7.

Histological damage was not reduced in the absence of interferon γ (IFN-γ) but was completely abolished in the absence of T cells. Damage score (A) and number of apoptotic bodies (B) of immunodeficient mice 24 hours and 48 hours after treatment with anti-CD3 control antibody. *p<0.05 compared with wild-type Balb/c mice, 24 hours (1) or 48 hours (2) after treatment with anti-CD3 antibody; NS, no significant difference compared with Balb/c mice, 24 hours (3) or 48 hours (4) after treatment with hamster HIgG (HIgG) control antibody (numbers indicate the number of animals treated).
Effect of single deficiency of the IFN-γ, TNF-α, Fas, or perforin mediated pathways
Serum levels of TNF-α were increased and peaked 30 minutes after anti-CD3. Serum IFN-γ was elevated at two hours and remained high until 12 hours after anti-CD3 (fig 6 ▶). Anti-CD3 Ab treatment also increased tissue TNF-α levels in the small intestine by one hour and this remained elevated at 20 hours (fig 6 ▶).
Figure 6.

Serum and small intestinal tissue cytokine levels in Balb/c mice injected with anti-CD3 antibody or hamster IgG (HIgG) control antibody. (A) Serum tumour necrosis factor α (TNF-α); (B) serum interferon γ (IFN-γ); (C) mucosal tissue TNF-α levels (n=3).
Tissue damage developed in a similar manner in IFN-γ deficient mice compared with Balb/c wild-type mice (fig 7 ▶). Signalling through the p55 TNF receptor, TNF-R1, is regarded as the main pathway by which TNF-α induces apoptosis in target cells. Treatment of TNF-R1−/− mice, TNF-R2−/−, or TNF-R1−/−×TNF-R2−/− mice, lacking both TNF receptors, with anti-CD3 antibody induced significant intestinal damage similar in severity to that seen in wild-type mice (fig 8 ▶). Also, the number of apoptotic bodies was not different compared with wild-type mice (fig 8 ▶). MRL lpr/lpr mice, which lack the Fas receptor, also developed diarrhoea and intestinal damage (fig 8 ▶). Even though the number of apoptotic bodies that were present 24 hours after treatment with anti-CD3 antibody was higher in MPJ background strain mice compared with Balb/c or C57BL/6 mice treated similarly, no significant difference in the number of apoptotic bodies between Fas deficient lpr mice and either of the wild-type strains was discernible (fig 8 ▶).
Figure 8.

T cell mediated histological damage and development of apoptotic bodies was partially reduced in the absence of perforin but not in the absence of Fas or tumour necrosis factor α (TNF-α) receptors. The score for small intestinal tissue damage (A), number of apoptotic bodies per five villus crypt units (B), and villus:crypt ratio (C) in mice 24 hours after treatment with anti-CD3 antibody are shown. No significant differences between tissue damage in anti-CD3 treated mice of the background strain and immunodeficient mice (MPJ for Fas deficient MRL lpr mice, C57Bl6 for all other mice) was detectable. Tissue damage was significantly reduced in anti-CD3 treated perforin deficient mice. *p<0.05 compared with C57Bl6 control mice treated with anti-CD3 antibody (1); NS, no significant difference compared with control C57Bl6 mice treated with anti-CD3 antibody (1) or control MPJ mice treated with anti-CD3 antibody (2) (numbers indicate the number of animals treated).
In contrast, the absence of perforin significantly reduced the severity of anti-CD3 induced enteropathy (fig 8 ▶). This was characterised by a significant reduction in the number of apoptotic bodies present in the epithelium 24 hours after anti-CD3 treatment although some villus architectural damage remained. Thus T cell induced tissue damage is partially mediated by perforin, while FasL and TNF-α pathways are not individually required.
Effect of combined deficiencies of TNF-α×Fas or perforin×FasL mediated pathways
In mice lacking Fas and both TNF receptors (TNF-R1×R2 lpr/lpr), mean tissue damage induced by T cell activation was not significantly reduced (fig 9 ▶). However, the number of apoptotic bodies was significantly reduced in these mice. Interestingly, in these double knockout mice, the reduction in villus:crypt ratio was not significantly different from that seen in wild-type mice treated with anti-CD3. Thus TNF-α and Fas are involved in the mediation of epithelial cell apoptosis but their combined presence is not required for alteration in mucosal architecture.
Figure 9.

Tumour necrosis factor α (TNF-α), Fas ligand (FasL), and perforin contribute to tissue damage. Tissue damage in anti-CD3 treated mice lacking Fas and both TNF-α receptors (TNF-R1×R2 lpr/lpr) was not statistically diminished (p=0.098). However, the number of apoptotic bodies was significantly reduced (p=0.012). Small intestinal damage was dramatically diminished in mice lacking both perforin and FasL (perforin−/− gld mice, p=0.001). No further increase in damage was seen at 48 hours post injection. FasL deficient mice remained susceptible to tissue damage (perforin+/− gld strain). The score for small intestinal tissue damage (A), number of apoptotic bodies per five villus crypt units (B), and villus:crypt ratio (C) in mice 24 hours after treatment with anti-CD3 antibody are shown. All mice to the right of the break in the X axis were treated with anti-CD3. *1, 2, 3, 4), p<0.05 compared with groups labelled with corresponding numbers; NS no significant difference compared with control C57Bl6 mice treated with anti-CD3 antibody (1) or control C57BL6 mice treated with HIgG antibody (2) (numbers indicate the number of animals treated).
In contrast, the histological damage score in mice deficient in both perforin and FasL (perforin−/−×gld) was significantly reduced relative to the wild-type strain (p=0.001). This damage was also statistically less severe than that seen in TNF-R1×R2 lpr/lpr mice or in perforin knockout mice (fig 9 ▶). Both changes in villus:crypt ratio and number of apoptotic bodies were significantly different in the perforin−/−×gld mice compared with control TNF-R1×R2 lpr/lpr mice as well as perforin knockout mice (fig 9 ▶). Furthermore, the immune mediated tissue damage in FasL deficient mice heterozygous for perforin deficiency (perforin+/−×gld) was not significantly reduced compared with wild-type mice, supporting the important role of perforin in mediating this damage. In perforin knockout×FasL deficient mice, histological tissue damage remained negligible 48 hours after in vivo T cell stimulation (p=0.032 compared with HIgG treated control mice) showing that the development of mucosal damage in these mice was not delayed. Thus FasL and perforin are the major contributors to the development of tissue damage induced by T cell activation while TNF-α appears to play a less significant role.
DISCUSSION
Activation of T cells is a pivotal step in the pathogenesis of inflammatory diseases of the bowel, such as coeliac disease, GvH disease, and the idiopathic inflammatory bowel diseases. Activated T cells participate in both regulatory and effector mechanisms involved in the development of an inflammatory response. The effector mechanisms are important for defence against microorganisms but may also cause significant bystander tissue damage. Mucosal flattening and enterocyte apoptosis have been recognised as hallmarks of T cell induced tissue injury in small intestinal inflammatory diseases.
To assess the contribution of T cell cytotoxicity to tissue damage that occurs in inflammatory bowel diseases, we established an in vivo mouse model of T cell mediated intestinal injury induced by direct in vivo activation of T cells via the common signal transduction moiety of the T cell receptor CD3. Administration of the anti-CD3 antibody 145-2C11 in vivo results initially in T cell activation followed by immunosuppression due to T cell depletion and downregulation of the T cell receptor.22,23 Injection of mice with anti-CD3 induced a severe small intestinal enteropathy that was characterised by epithelial crypt cell apoptosis and villus flattening. This supports the notion that villus flattening and enterocyte apoptosis can result from T cell activation.24
In the large intestine, the structural sequelae of T cell activation were less obvious. A decreased sensitivity towards proapoptotic stimuli has been suggested for large intestinal epithelial stem cells that usually express bcl-2, in contrast with small intestinal stem cells, which are bcl-2 negative and rapidly express bax on cellular damage. Knockout studies have demonstrated that both p53 and Bcl2 are important regulators of stress induced apoptosis but that there are significant differences between early and late time points. The cumulative effect of stress induced apoptosis on tissue architecture is not straightforward, and cell cycle arrest also plays a critical role.25 Our data suggest that although apoptotic bodies were seen in the colon of anti-CD3 mice, the tissue damage was minimal, perhaps reflecting less dependence of the colonic epithelium on rapid epithelial cell turnover. Alternatively, the differences between small and large bowel damage may be related to a differential response of local T cells to activation signals.26 We assessed in vivo activation of T cells by measuring BrdU incorporation as a reflection of cell division. The increase in BrdU staining of IEL from mice treated with anti-CD3 antibody may be due to altered in vivo proliferative response of activated/memory T cells found in mucosal tissues27 or alternatively an altered cytokine environment within the mucosa. None the less, detection of T cell activation in the mucosa suggests that the anti-CD3 antibody can actually activate mucosal T cells in vivo. It remains to be shown which mucosal compartment of T cells is actually mediating the mucosal damage.
We investigated the role of distinct pathways of T cell mediated cytotoxicity in the induction of intestinal damage. In vitro experiments suggest that T cell induced target cell damage, including apoptosis of epithelial cells, is mediated by TNF-α, FasL, and perforin.28 TNF-α is a central mediator of intestinal inflammation and is increased in human inflammatory bowel disease. Our results indicate that the absence of TNF-α pathways failed to prevent T cell induced intestinal damage. This was not anticipated given that TNF-α is toxic for epithelial cells in vitro10,29 and in vivo administration of TNF-α induces an enteropathy characterised by villus shortening.9,30 Furthermore, TNF-α induces the release of matrix metalloproteinases that digest connective tissue and is involved in the remodelling of the mucosa in the in vitro fetal intestinal explant model of T cell mediated injury.17,31 On the other hand, activated T cells from inflamed mucosa of interleukin 2 deficient mice produced significant amounts of TNF-α and IFN-γ but were unable to kill or reduce the growth of syngeneic intestinal epithelial cells.32 Lastly, anti-TNF therapy is only effective in a subgroup of patients with Crohn's disease, supporting the notion that other potent mediators of tissue damage contribute to immune mediated mucosal damage.33
The role of Fas/FasL and perforin in inflammatory diseases of the bowel has only recently been studied. Increased expression of FasL has been detected in inflammatory bowel disease,3,12 GvH disease,34,35 and coeliac disease,36 and treatment with anti-FasL Ab was therapeutic in a murine model of GvH disease.14 On the other hand, the absence of FasL failed to prevent inflammation in a mouse model of colitis induced by transfer of bone marrow cells into Tg26 mice.37 Our data indicate that the Fas/FasL pathway was not required for T cell induced tissue damage or the development of apoptosis.
Perforin on the other hand played an important role in T cell induced mucosal damage. Perforin expression is increased in GvH disease,38–40 coeliac disease,36 and murine models of inflammatory bowel disease.41 Perforin contributes to apoptosis by facilitating the introduction of granzymes into the target cell via the perforin pore and increased levels of granzyme B mRNA have been detected in cells adjacent to apoptotic epithelial cells in inflamed Crohn's disease tissue.42 Furthermore, experiments with the Tg26 transfer model of colitis indicate that the absence of perforin reduced tissue damage.37
In the absence of both TNF-α receptors and Fas (TNF-R1×R2−/− lpr/lpr mice), loss of villi occurred in spite of a reduction in the number of apoptotic cells. In the absence of perforin and FasL, both loss of villus structure and apoptotic bodes were decreased. This suggests that in the presence of proapoptotic mediators and perforin, epithelial damage occurs mainly through apoptosis while perforin may also contribute to non-apoptotic mechanisms of target cell damage such as pore mediated lysis of target cells in the absence of TNF-α and Fas/FasL.
Therefore, a number of cooperative pathways contribute to the induction of apoptosis and mucosal damage during T cell induced enteropathy. This is supported by in vitro studies showing that T cell induced target cell damage, including apoptosis of epithelial cells, is mediated by the combined effect of TNF-α, FasL, and perforin.28 The contribution of Fas/FasL is underestimated if cooperative effects between cytotoxic effector mechanisms are not considered. Absence of Fas/FasL alone failed to reduce apoptosis but the combined absence of both TNF-α and Fas significantly reduced enterocyte apoptosis. Therefore, even though TNF-α and FasL are both not individually required for the development of mucosal damage in our model, both molecules contribute to induction of apoptosis. Similarly, the Fas/FasL and perforin pathways combine to induce even greater epithelial cell apoptosis and mucosal damage. Similar results were obtained in studies of T cell induced tissue destruction in intestinal inflammatory disease36 and GvH disease models.38,40
On the other hand, our finding that TNF-α receptor and IFN-γ deficiency alone did not reduce tissue damage should also to be interpreted with caution. The potential remains for these cytokines to cooperate with other cytotoxic mediators. None the less, the role of TNF-α and FasL is outweighed by perforin mediated killing. It remains possible therefore that in the absence of perforin, the effects of TNF-α and IFN-γ would be more apparent.
In conclusion, the experimental enteropathy induced by in vivo anti-CD3 antibody injection in mice demonstrates that multiple pathways of cytotoxicity, including TNF-α, Fas/FasL, and particularly perforin, mediate apoptosis and are associated with architectural damage. These results highlight the redundancy of cytotoxic T cell mechanisms and predict that inhibiting any one pathway is unlikely to prevent all aspects of immunopathology associated with T cell induced enteropathies.
Acknowledgments
We would like to thank Dr Jacques Peschon, Immunex Corporation, for supplying the TNF-R2−/− and TNF-R1×R2 deficient mice and Dr Tak Mak, AMGEN Institute, Toronto for the TNF-R1−/− mice. The study was funded by the Medical Research Council of Canada and the Crohn's and Colitis Foundation of Canada. KC holds an Ontario Minister of Health Career Scientist Award. MM was supported by a fellowship from the Deutsche Forschungsgemeinschaft (grant Me 1403/1-1). ERP is supported by NIH grant Nos CA39201, CA 57904, CA 80228, and DOD grant No DAMD517-98-1-8317.
Abbreviations
Ab, antibody
BrdU, bromodeoxyuridine
CsA, cyclosporin A
Dex, dexamethasone
GvH, graft versus host
HIgG, hamster IgG
IFN-γ, interferon γ
TNF, tumour necrosis factor
FasL, Fas ligand
PBS, phosphate buffered saline
TUNEL, terminal uridine nick-end labelling
IEL, intraepithelial lymphocytes
REFERENCES
- 1.MacDonald TT, Spencer J. Evidence that activated mucosal T cells play a role in the pathogenesis of enteropathy in human small intestine. J Exp Med 1988;167:1341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moss SF, Attia L, Scholes JV, et al. Increased small intestinal apoptosis in coeliac disease. Gut 1996;39:811–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strater J, Wellisch I, Riedl S, et al. CD95 (APO-1/Fas)-mediated apoptosis in colon epithelial cells: a possible role in ulcerative colitis. Gastroenterology 1997;113:16. [DOI] [PubMed] [Google Scholar]
- 4.Yang X, Stennicke HR, Wang B, et al. Granzyme B mimics apical caspases. Description of a unified pathway for trans-activation of executioner caspase-3 and -7. J Biol Chem 1998;273:34278–83. [DOI] [PubMed] [Google Scholar]
- 5.Thiele DL, Eigenbrodt ML, Bryde SE, et al. Intestinal graft-versus-host disease is initiated by donor T cells distinct from classic cytotoxic T lymphocytes. J Clin Invest 1989;84:1947–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baker MB, Altman NH, Podack ER, et al. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J Exp Med 1996;183:2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lake-Bullock MH, Bryson JS, Jennings CD, et al. Inhibition of graft-versus-host disease—Use of a T cell-controlled suicide gene. J Immunol 1997;158:5079–82. [PubMed] [Google Scholar]
- 8.Brown GR, Lindberg G, Meddings J, et al. Tumor necrosis factor inhibitor ameliorates murine intestinal graft-versus-host disease. Gastroenterology 1999;116:593–601. [DOI] [PubMed] [Google Scholar]
- 9.Guy-Grand D, DiSanto JP, Henchoz P, et al. Small bowel enteropathy: role of intraepithelial lymphocytes and of cytokines (IL-12, IFN-gamma, TNF) in the induction of epithelial cell death and renewal. Eur J Immunol 1998;28:730–44. [DOI] [PubMed] [Google Scholar]
- 10.Deem RL, Shanahan F, Targan SR. Triggered human mucosal T cells release tumour necrosis factor-alpha and interferon-gamma which kill human colonic epithelial cells. Clin Exp Immunol 1991;83:79–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abreu-Martin MT, Palladino AA, Faris M, et al. Fas activates the JNK pathway in human colonic epithelial cells: lack of a direct role in apoptosis. Am J Physiol 1999;276:G599–605. [DOI] [PubMed] [Google Scholar]
- 12.Ueyama H, Kiyohara T, Sawada N, et al. High Fas ligand expression on lymphocytes in lesions of ulcerative colitis. Gut 1998;43:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakai T, Kimura Y, Inagaki-Ohara K, et al. Fas-mediated cytotoxicity by intestinal intraepithelial lymphocytes during acute graft-versus-host disease in mice. Gastroenterology 1997;113:168–74. [DOI] [PubMed] [Google Scholar]
- 14.Miwa K, Hashimoto H, Yatomi T, et al. Therapeutic effect of an anti-Fas ligand mAb on lethal graft-versus-host disease. Int Immunol 1999;11:925–31. [DOI] [PubMed] [Google Scholar]
- 15.Maggi CA. Capsaicin and primary afferent neurons: from basic science to human therapy? J Auton Nerv Syst 1991;33:1–14. [DOI] [PubMed] [Google Scholar]
- 16.Nakajima S, Krishnan B, Ota H, et al. Mast cell involvement in gastritis with or without Helicobacter pylori infection. Gastroenterology 1997;113:746–54. [DOI] [PubMed] [Google Scholar]
- 17.Pender SLF, Tickle SP, Docherty AJP, et al. A major role for matrix metalloproteinases in T cell injury in the gut. J Immunol 1997;158:1582–90. [PubMed] [Google Scholar]
- 18.Spielman J, Lee RK, Podack ER. Perforin/Fas-ligand double deficiency is associated with macrophage expansion and severe pancreatitis. J Immunol 1998;161:7063–70. [PubMed] [Google Scholar]
- 19.Hirsch R, Eckhaus M, Auchincloss H Jr, et al. Effects of in vivo administration of anti-T3 monoclonal antibody on T cell function in mice. I. Immunosuppression of transplantation responses. J Immunol 1988;140:3766–72. [PubMed] [Google Scholar]
- 20.Croitoru K, Bienenstock J, Ernst PB. Phenotypic and functional assessment of intraepithelial lymphocytes bearing a “forbidden” α/β T cell receptor. Int Immunol 1994;6:1467–73. [DOI] [PubMed] [Google Scholar]
- 21.Ansell JD, Bancroft GJ. The biology of the SCID mutation. Immunol Today 1989;10:322. [DOI] [PubMed] [Google Scholar]
- 22.Leo O, Foo M, Sachs DH, et al. Identification of a monoclonal antibody specific for a murine T3 polypeptide. Proc Natl Acad Sci USA 1987;84:1374–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith JA, Bluestone JA. T cell inactivation and cytokine deviation promoted by anti-CD3 mAbs. Curr Opin Immunol 1997;9:648–54. [DOI] [PubMed] [Google Scholar]
- 24.Lionetti P, Breese E, Braegger CP, et al. T-cell activation can induce either mucosal destruction or adaptation in cultured human fetal small intestine. Gastroenterology 1993;105:373–81. [DOI] [PubMed] [Google Scholar]
- 25.Watson AJ, Pritchard DM. Lessons from genetically engineered animal models. VII. Apoptosis in intestinal epithelium: lessons from transgenic and knockout mice. Am J Physiol Gastrointest Liver Physiol 2000;278:G1–5. [DOI] [PubMed] [Google Scholar]
- 26.Camerini V, Panwala C, Kronenberg M. Regional specialization of the mucosal immune system. Intraepithelial lymphocytes of the large intestine have a different phenotype and function than those of the small intestine. J Immunol 1993;151:1765–76. [PubMed] [Google Scholar]
- 27.Tough DF, Sun S, Zhang X, et al. Stimulation of naive and memory T cells by cytokines. Immunol Rev 1999;170:39–47. [DOI] [PubMed] [Google Scholar]
- 28.Lee RK, Spielman J, Zhao DY, et al. Perforin, Fas ligand, and tumor necrosis factor are the major cytotoxic molecules used by lymphokine-activated killer cells. J Immunol 1996;157:1919–25. [PubMed] [Google Scholar]
- 29.Sveinbjornsson B, Rushfeldt C, Olsen R, et al. Cytotoxic effect of cytokines on murine colon carcinoma cells involves TNF-mediated apoptosis. Biochem Biophys Res Commun 1997;233:270. [DOI] [PubMed] [Google Scholar]
- 30.Garside P, Bunce C, Tomlinson RC, et al. Analysis of enteropathy induced by tumour necrosis factor alpha. Cytokine 1993;5:24–30. [DOI] [PubMed] [Google Scholar]
- 31.Pender SLF, Fell JME, Chamow SM, et al. A p55 TNF receptor immunoadhesin prevents T cell-mediated intestinal injury by inhibiting matrix metalloproteinase production. J Immunol 1998;160:4098–103. [PubMed] [Google Scholar]
- 32.Baumgart DC, Olivier WA, Reya T, et al. Mechanisms of intestinal epithelial cell injury and colitis in interleukin 2 (IL2)-deficient mice. Cell Immunol 1998;187:52–66. [DOI] [PubMed] [Google Scholar]
- 33.Targan SR, Hanauer SB, Van Deventer et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn's disease. Crohn's Disease cA2 Study Group. N Engl J Med 1997;337:1029–35. [DOI] [PubMed] [Google Scholar]
- 34.Sakai T, Kimura Y, Inagaki-Ohara K, et al. Fas-mediated cytotoxicity by intestinal intraepithelial lymphocytes during acute graft-versus-host disease in mice Fas-mediated cytotoxicity by intestinal intraepithelial lymphocytes during acute graft-versus-host disease in mice. Gastroenterology 1997;113:168–74. [DOI] [PubMed] [Google Scholar]
- 35.Shustov A, Nguyen P, Finkelman F, et al. Differential expression of Fas and Fas ligand in acute and chronic graft-versus-host disease: Up-regulation of Fas and Fas ligand requires CD8+ T cell activation and IFN-gamma production. J Immunol 1998;161:2848–55. [PubMed] [Google Scholar]
- 36.Ciccocioppo R, Di Sabatino A, Parroni et al. Cytolytic mechanisms of intraepithelial lymphocytes in coeliac disease (CoD). Clin Exp Immunol 2000;120:235–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simpson SJ, De Jong YP, Shah et al. Consequences of Fas-ligand and perforin expression by colon T cells in a mouse model of inflammatory bowel disease. Gastroenterology 1998;115:849–55. [DOI] [PubMed] [Google Scholar]
- 38.Graubert TA, DiPersio JF, Russell JH, et al. Perforin/granzyme-dependent and independent mechanisms are both important for the development of graft-versus-host disease after murine bone marrow transplantation. J Clin Invest 1997;100:904–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pham CT, Ley TJ. The role of granzyme B cluster proteases in cell-mediated cytotoxicity. Semin Immunol 1997;9:127–33. [DOI] [PubMed] [Google Scholar]
- 40.Braun MY, Lowin B, French L, et al. Cytotoxic T cells deficient in both functional Fas ligand and perforin show residual cytolytic activity yet lose their capacity to induce lethal acute graft-versus-host disease. J Exp Med 1996;183:657–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aarvak T, Chabaud M, Miossec P, et al. IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol 1999;162:1246–51. [PubMed] [Google Scholar]
- 42.Jenkins D, Seth R, Kummer JA, et al. Differential levels of granzyme B, regulatory cytokines, and apoptosis in Crohn's disease and ulcerative colitis at first presentation. J Pathol 2000;190:184–9. [DOI] [PubMed] [Google Scholar]