

Abstract
Aims: Intracellular folate deficiency has been implicated in colonic carcinogenesis in epidemiological studies and animal and human cancer models. Our aim was to determine the effect of folate supplementation on patients with recurrent adenomatous polyps using rectal mucosal cell proliferation as a biomarker.
Patients and methods: Eleven patients with recurrent adenomatous polyps of the colon were randomised into a treatment group (n=6) receiving a dietary supplement of 2 mg folic acid per day for three months and a control group (n=5) receiving a placebo. Rectal biopsies where taken at 10 cm from the anal verge prior to supplementation and repeated at four, 12, and 18 weeks from the start of the supplementation. Each biopsy was immediately incubated in culture medium enriched with bromodeoxyuridine (BrdU). The S phase cells which incorporated BrdU into their DNA were identified following immunohistochemical staining. Twenty five orientated crypts were identified for each time point and the number and position of BrdU positive and BrdU negative cells were counted. BrdU labelling indices (LIs) were calculated for the entire crypt and for each of five equal compartments running consequently from the base to the luminal surface.
Results: The LI of the treatment group (9.1 (6.7, 12.3)) and the control group (9.3 (7.8, 10.3)) were comparable at the start. Over the duration of the supplementation period, LI in the control group did not alter significantly (9.3 (7.8, 10.3) v 9.6 (8.9, 10.4)). However, LI of the folate treated group was lowered after 12 weeks of supplementation (9.1 (6.7, 12.3) v 7.4 (5.3, 9.6)). Analysis of the LI for compartments within the crypt showed that the most significant drop in number of proliferating cells was in the upper most regions of the crypt.
Conclusion: These data indicate that (a) folate supplementation decreases colonic mucosal cell proliferation in a high risk group for colon cancer and (b) the most significant reduction takes place at the luminal aspect of the crypt.
Keywords: adenomatous polyps, chemoprevention, folate, mucosal cell proliferation
The role of diet in the prevention of colorectal cancer (CRC) has received a great deal of attention. Investigations into the possible effect of folate intake on colorectal neoplasia have been stimulated by the observations that hypomethylation of DNA appears to be an early step in colorectal carcinogenesis1–3 and that low intake of fruit and vegetables are positively associated with colon cancer.4 This together with reports that certain fruits and vegetables may be protective against CRC through mechanisms other than their fibre content5 has resulted in renewed interest into the mechanisms involved in dietary chemoprevention.
Several studies have reported an association between dietary folate intake and development of adenomatous polyps and CRC.6–16
In animal models of CRC, folate deficiency has been found to enhance the development of colonic neoplasia.17 Conversely, dietary supplementation, up to four times the required daily amount, protects against the development of macroscopic colonic neoplasms.18
To avoid the use of costly long term follow up studies, researchers examining the potential of dietary agents have validated measurements of gastrointestinal cell proliferation which can be employed as intermediate biomarkers of colorectal cancer. In vitro incorporation of BrdU into the DNA of cells allows identification of the cells in the S phase of the cycle which represent the actively proliferating cells. This has been shown to be a reliable and reproducible method which has been used successfully in several chemoprevention trials.19,20 Furthermore, there is evidence that cell proliferation of the rectal mucosa is representative of the entire colon.21–25
The randomised double blind study described here is the first of its kind to evaluate the effect of dietary supplementation of folate on the proliferative pattern of rectal mucosa in high risk patients with recurrent adenomatous colorectal polyps.
PATIENTS AND METHODS
Twenty patients with recurrent adenomatous polyps of the colon were selected for inclusion in this study. Exclusion criteria were: (1) previous history of CRC; (2) family history of colon cancer or familial polyposis; (3) current metabolic or life threatening disease; (4) recent use of vitamin supplements or non-steroidal anti-inflammatory drugs; (5) pregnancy; and (6) anaemia or B12 deficiency. Patients were randomised into two groups, with 10 in each group (five males, five females). Informed consent was obtained from each patient before inclusion. Ethics approval was granted by Queen's University of Belfast Research Ethical Committee.
All patients received a 12 week supply of tablets. The test group received 2 mg per day of folic acid whereas the control group received a placebo tablet of sodium chloride. Each patient had rigid sigmoidoscopy performed without previous bowel preparation and four biopsies were taken from the rectal mucosa at 10 cm from the anal verge. This sampling was performed at the outset of the study, at four weeks, 12 weeks, and 18 weeks from the start. One biopsy was sent for histopathological examination and any evidence of mucosal inflammation, atypia, or dysplasia resulted in exclusion from the study.
Compliance
Compliance was assessed by pill count. Subjects were provided with three bottles containing the exact number of pills plus three extra.
Blood sampling
Blood samples were taken from each patient at each of the four visits and used to measure red blood cell levels of folate. Red blood cell levels of folate were measured as described in the manual with the reagents supplied (SimulTRAC-S Radioassay Kit; ICN Pharmaceuticals, New York, USA). This work was carried out at the clinical chemistry laboratories in Belfast City Hospital (Lisburn Road, Belfast, UK).
Dietary questionnaire
To evaluate any alteration in diet which may have taken place following interview, patients were asked to complete a three day dietary questionnaire at commencement of the study and at the end of the supplementation period.26 Patients were asked to record their total intake during three separate 24 hour periods, usually two week days and one weekend day. Patients were instructed about the information required—that is, the type and amount of food and drink consumed, and how the food and drink were prepared. From this information and using food analysis (The composition of foods, 5th edition, McCance and Widdowson's) the information was entered into a computer (Microdiet software; University of Salford, Salford, UK) which was able to calculate the total calorific intake, along with breakdown into each component of the food—that is, fat, carbohydrate, proteins, and vitamins with trace elements.
Bromodeoxyuridine incorporation
Biopsies were divided into 1 mm3 pieces and any mucus or faecal residue was detached. Biopsies were orientated under a dissecting microscope onto nitrocellulose paper with the velvety side facing upwards so that the submucosa was next to the paper with the crypts uppermost. They were immediately placed in a wire basket and suspended above a gas filter bubbling 95% O2 and 5% CO2, immersed under 30 ml of Dulbecco's modified Eagle's medium with 3 ml of fetal calf serum (10%), 360 μl l-glutamine (200 mM), and 1830 μl of stock bromodeoxyuridine (BrdU, final concentration 1000 μM). Incubation was continued for 60 minutes in a water bath at a constant temperature of 37°C.
Following this, samples were fixed in 70% ethanol for four hours. They were then processed routinely to paraffin wax on an automatic tissue processor. Then, biopsies were carefully embedded individually and orientated with the paper standing on its edge in paraffin wax. Sections (3 μm) were cut from the paraffin blocks at a minimum distance of 40 μm apart and dried overnight at room temperature before immunohistochemical staining.
Sections were dewaxed in xylene and rehydrated through descending concentrations of alcohol. Slides were thoroughly washed in phosphate buffered saline (PBS, pH 7.1). DNA was denatured in 1 M hydrochloric acid at 37°C for 12 minutes to expose the incorporated BrdU in single stranded DNA and inhibit endogenous peroxidase activity.
Bromodeoxyuridine immunohistochemistry
Primary antibody solution (100 μl) containing 4 μl human serum and 2 μl mouse anti-BrdU (Bu20a) monoclonal antibody (Dako M744, Bucks, UK) diluted in 1:50 PBS with 0.05% Tween 20 (Sigma P1379) was applied to each slide and incubated for 60 minutes at room temperature. This was followed by 100 μl of biotinylated secondary antibody solution, containing 4 μl human serum and 0.5 μl biotinylated rabbit anti-mouse F(ab`)2 antibody (Dako E413) diluted to 1:200 in PBS with Tween, for a further 60 minutes. Slides were rinsed in PBS before 100 μl streptavidin-biotin-peroxidase complex (Dako K 377) was applied for 30 minutes. Antibody binding was visualised with the chromogen diaminobenzidine (Sigma, Dorset, UK), primed with 100 μl of 30% H2O2. Finally, the sections were lightly counterstained with Harris haematoxylin (Sigma HHS-128) for 10 seconds to allow the non-labelled nuclei to be visualised and counted. As a negative control, sections were incubated with a class matched non-specific mouse Ig monoclonal antibody instead of the primary Bu20a monoclonal antibody. As a positive control, a section known to stain positively was included in each run.
Calculation of the labelling index in mucosal samples
Using direct microscopy, well orientated longitudinally sectioned crypts that could be identified from luminal surface to muscularis mucosae were used for analysis. To facilitate scoring, each crypt was divided at the base into two crypt columns or hemicrypts. Starting at the base of the hemicrypt, cells were numbered up to the luminal surface of the colon to determine the number of cells per hemicrypt and then divided into five equal compartments each containing a fifth of the total number of cells. Twenty five complete crypts (50 hemicrypts) were examined for each patient at each time point. The number and position of BrdU labelled cells in the hemicrypt were recorded. The labelling index (LI) was determined for the whole hemicrypt and for each compartment, by dividing the number of labelled cells by the total cells and multiplying by 100.
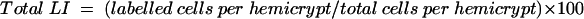 |
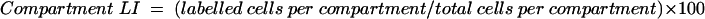 |
Statistical analysis
All LI data obtained throughout the study displayed a normal distribution. On this basis, a paired Student's t test was used to identify differences between baseline LIs and LIs at 12 weeks in the control and folate groups separately. A two sample t test was utilised to detect differences in mean reduction over the supplementation period between the treated and control groups. Due to the small sample size, the analysis was also performed using Wilcoxon's signed rank test for comparison between groups and the Mann-Whitney test for paired analysis to corroborate the findings.
The results of red blood cell levels of folate and dietary nutrient intake were analysed using a non-parametric method (Mann-Whitney U test). In all cases, results were considered to be significant when p<0.05.
RESULTS
Both groups were similar with respect to sex distribution, age, and weight.
Compliance
Only 11 patients completed the trial; the main reason for non-compliance was poor tolerance for repeated rigid sigmoidoscopy. All patients returned three bottles which contained the supplements. In only two cases was there less than three pills remaining in each bottle.
Blood sampling
As expected, there was a marked increase in mean red cell folate levels in the folate supplemented group (from 253 μg/l before (t=0 weeks) to 653 μg/l after treatment (t=12 weeks; p<0.05). The control group showed no change in mean red cell levels of folate (198 μg/l before (t=0 weeks) and 200 μg/l after treatment period (t=12 weeks)).
Dietary questionnaire
Mean daily food intake at the start of the study and at the end of 12 weeks in the two groups were similar and there was no alteration in the intake of folate through dietary means between the two time points (table 1 ▶).
Table 1.
Comparison of dietary patterns between the two groups before and after intervention
| Before intervention | After intervention | |||
| Daily intake | Control (n=5) | Folate (n=6) | Control (n=5) | Folate (n=6) |
| Energy (kcal) | 1625 (380) | 1802 (286) | 1657 (617) | 1692 (327) |
| Protein (g) | 67 (7) | 68 (12) | 64 (17) | 65 (18) |
| Fat (g) | 61 (26) | 58 (19) | 67 (27) | 56 (14) |
| Carbohydrate (g) | 196 (50) | 191 (46) | 199 (76) | 179 (51) |
| Folate (μg) | 170 (39) | 172 (58) | 193 (106) | 197 (69) |
Values are mean (SD).
Labelling index
While crypt length was not measured, the total number of cells per crypt were counted. The total number of cells per crypt were similar between the controls and folate treated group (60 (6.4) and 63 (7.1), respectively) (mean (SD)). The LI for the two groups was comparable before intervention. After 12 weeks of supplementation there was no significant change in the LI of the control group but there was a significant decrease in the LI of the folate treated group (p=0.044) (table 2 ▶). The mean reduction in total LI did reach significance after 12 weeks of supplementation when comparing the treated and control groups (p=0.05, two sample t test).
Table 2.
Comparison of the labelling index (LI) in the two treatment groups over the four time points (supplementation was given only for the first 12 weeks)
| Time (weeks) | ||||
| Group | 0 | 4 | 12 | 18 |
| Controls | 9.3 (6.7, 12.3) | 9.0 (7.7, 10.5) | 9.6 (8.9, 10.4) | 9.1 (8.5, 9.8) |
| Folate | 9.1 (7.8, 10.3) | 8.3 (6.2, 10.5) | 7.4 (5.3, 9.6)*† | 8.0 (6.7, 9.5) |
*p<0.044 (paired t test), †p=0.03 (Wilcoxon's signed rank test).
Values are mean patient LI (95% confidence limits).
There was a decrease in LI after four weeks of supplementation with folic acid but this only reached statistical significance after 12 weeks.
When the data were analysed on an individual patient basis it was evident that the LI for the crypt and each individual compartment in the control group did not alter significantly throughout the duration of the study. In the folate treated group, although the overall group LI was significantly lower after 12 weeks of supplementation, the total crypt LI was not decreased to the same extent in each of the patients (fig 1 ▶). When individual compartment LIs were examined, there was no change in the LI of compartments at the base of the crypt (compartments 1, 2, and 3) whereas the LI of the compartments at the luminal surface of the crypts (compartments 4 and 5) was reduced significantly in the folate treated group after 12 and 18 weeks supplementation (p=0.006 and 0.007 (paired t test), respectively) (fig 2 ▶, table 3 ▶). There was no change in compartmental LIs of the control group. When the mean reduction in total LI of compartments 4 and 5 was analysed, there was again a significant difference between the two groups (p=0.01, two sample t test). It was also noted that the LI for the entire crypt and the individual compartments showed a trend towards their pretreatment values after supplementation had been discontinued for six weeks (figs 1, 2 ▶ ▶).
Figure 1.

Total crypt labelling indices (LI) for each patient in the control group (A) and folate group (B). Each line connects the LIs at each of the four examination points.
Figure 2.

Upper crypt (compartments 4 and 5) labelling indices (LI) for each patient in the control group (A) and folate group (B). Each line connects the LIs at each of the four examination points.
Table 3.
Comparison of the labelling index (LI) (compartments 4 and 5) in the two treatment groups over the four time points (supplementation was given only for the first 12 weeks)
| Time (weeks) | ||||
| Group | 0 | 4 | 12 | 18 |
| Controls | 3.1 (1.4, 4.7) | 2.4 (1.5, 3.3) | 2.6 (2.0, 3.2) | 2.0 (1.5, 2.5) |
| Folate | 3.9 (2.1, 5.7) | 2.6 (0.7, 4.5) | 0.6 (0, 1.6)* | 1.5 (0.6, 2.4)** |
*p=0.006, **p=0.007 (paired t test).
Values are mean patient LI (compartments 4 and 5) (95% confidence limits).
DISCUSSION
This study has demonstrated that folate supplementation modulates the state of proliferative cells in the rectal mucosa. More importantly, it demonstrates that the reduction in cell proliferation is induced mainly at the luminal aspect of the crypt. The presence of labelled cells in the upper crypt is thought to reflect defective cell proliferation control and delayed onset of normal differentiation. In fact, it has been commented that evaluation of the upper most compartments of the crypt provides the most discriminating evidence of high risk.19,22,27 Furthermore, studies in animal models have shown that nutrient induced changes in colonic mucosal crypt cells are directly related to changes in tumour risk.28,29 In 1997, Biasco and colleagues demonstrated that folate supplementation regulated rectal mucosal cell proliferation in patients with long standing ulcerative colitis.30 However, this is the first report of the effect of folate in patients with recurrent adenomatous polyps.
Both the dietary questionnaires and red cell folate results provide evidence that other environmental factors have been controlled as far as possible. This lends support to our finding that the effect seen in the proliferative pattern is produced by folate supplementation. Studies of blood folate levels in relation to colorectal adenomas raise the question as to whether patients with cancer or polyps might have less efficient absorption and/or metabolism of folate than normal. Interestingly, mean red blood cell folate levels of adenoma patients have been reported to be substantially less than those of controls.12,13 It is accepted that the usual dietary intake of folate is about 200 μg/day,12,31 therefore we used a supratherapeutic dose of 2 mg/day to achieve a significant increase in body folate levels, as shown by the red cell measurements. There is a report that increased dietary folate of up to four times the basal requirement in an animal model demonstrates a beneficial effect in reducing macroscopic tumour load.18 Interestingly, it was found that dietary means of increasing daily folate intake was only related to a modest reduction in the risk of colon cancer while the reduction was more pronounced with specific folate supplements.8 Conversely, there are risks associated with folate supplementation, especially in patients with B12 deficiency, and interestingly in subjects with advanced malignancy. In these patients increased folate levels are believed to increase malignant cell turnover. There is also a concern that subjects who are receiving antiepileptic medication may need to alter the dosage of their drugs during folate supplementation. However, there is no evidence that folate supplementation interferes with the efficiency of antifolate chemotherapeutic agents. Timing of any dietary supplementation is also an important consideration because this may influence whether the effects suppress or enhance tumorigenesis.28
There are a number of hypotheses to explain the putative relationship between low intake of folate and colon cancer, which may be reversed by increasing total body folate level by dietary means or supplementation. These include: (a) induction of DNA hypomethylation; (b) induction of DNA damage; (c) impairment of DNA repair; (d) secondary depletion of choline; and (e) chromosomal abnormalities at fragile sites.
In the presence of low levels of folate, DNA hypomethylation may be induced in colorectal neoplasia and occurs as a global phenomenon.32 DNA hypomethylation is noted to increase from mucosa in normal subjects to normal mucosa in subjects with neoplastic lesion, to adenomas, and finally carcinoma.33,34 DNA hypermethylation precedes most of the other molecular events that occur in colorectal carcinogenesis, suggesting that alterations in DNA methylation are aetiologically important.
Folate deficiency may also induce DNA strand breaks which are associated with neoplastic transformation.35 Supraphysiological levels of folate supplementation (four times the daily dietary requirement) lead to a degree of p53 integrity greater than that observed with basal diet.35 When human lymphoid cells are treated with inhibitors of folate metabolism, such as methotrexate, folate dependent thymidine synthesis is impaired, leading to substitution of thymidylate by uridylate in DNA synthesis.36 This may be a mechanism whereby low folate levels lead to the occurrence of missense or non-sense gene mutations.
Folate deficiency may impair DNA repair in the colonic mucosa. In male Sprague-Dawley rats, folate deficiency reduces DNA excision repair.37 It does not itself affect mismatch repair, as measured by microsatellite instability. The established rat colon procarcinogen 1,2-dimethylhydrazine does not induce microsatellite instability on its own but does induce this when animals are folate deficient. Thus folate deficiency may diminish multiple aspects of DNA repair in the colonic mucosa.
Another mechanism of action may involve activation of protein kinase C, which is associated with CRC in humans and animals. This may occur because of a deficiency in choline which, like methionine, is a methyl donor. In cases of long term folate deficiency, choline levels are also reduced.38
Folate deficiency may also lead to chromosomal abnormalities at fragile sites. CRC has been associated with disruption of chromosomal integrity with loss of tumour suppresser gene activity.39 It may also lead to impaired ability of natural killer cells to destroy dysplastic or neoplastic cells.40
In conclusion, these data provide evidence that folate supplementation can regulate colonic mucosa cell proliferation in patients with recurrent adenomatous polyps, as a subgroup at high risk of colon cancer. The reduction in the proliferative cell numbers is most significant at the upper zones of the crypts. These data also indicate that the effect of the supplementation may persist for a period after the supplement is discontinued. This report highlights the need for further investigation on the role of folate as a chemopreventative agent in patients at risk of colon cancer.
Acknowledgments
This work was supported by Research and Development Grant (DHSS), Royal College of Surgeons of Edinburgh, and Action Cancer (Northern Ireland).
Abbreviations
BrdU, bromodeoxyuridine
LI, labelling index
CRC, colorectal cancer
PBS, phosphate buffered saline
REFERENCES
- 1.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301:89–91. [DOI] [PubMed] [Google Scholar]
- 2.Goelz SE, Vogelstein B. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 1985;228:187–90. [DOI] [PubMed] [Google Scholar]
- 3.Slattery ML, Potter JD, Duncan DM, et al. Dietary fats and colon cancer: assessment of risk associated with specific fatty acids. Int J Cancer 1997;73:670–7. [DOI] [PubMed] [Google Scholar]
- 4.Willet W. The search for the cause of breast and colon cancer. Nature 1989;338:389–94. [DOI] [PubMed] [Google Scholar]
- 5.Le Marchand L, Hankin JH, Wilkens LR, et al. Dietary fibre and colorectal cancer risk. Epidemiology 1997;8:658–65. [DOI] [PubMed] [Google Scholar]
- 6.Benito E, Stiggelbout A, Bosch FX, et al. Nutritional factors in colorectal cancer risk: a case-control study in Majorca. Int J Cancer 1991;49:161–7. [DOI] [PubMed] [Google Scholar]
- 7.Benito E, Cabeza E, Moreno V, et al. Diet and colorectal adenomas: a case-control study in Majorca. Int J Cancer 1993;55:213–19. [DOI] [PubMed] [Google Scholar]
- 8.Giovannucci E, Stampfer MJ, Colditz GA, et al. Multivitamin use, folate, and colon cancer in women in the Nurse's Health Study. Ann Intern Med 1998;129:517–24. [DOI] [PubMed] [Google Scholar]
- 9.Meyer F, White E. Alcohol and nutrients in relation to colon cancer in middle-aged adults. Am J Epidemiology 1993;138:225–36. [DOI] [PubMed] [Google Scholar]
- 10.Giovannucci E, Rimm EB, Ascherio A, et al. Alcohol, low-methionine-low-folate diets, and risk of colon cancer in men. J Natl Cancer Inst 1995;87:265–73. [DOI] [PubMed] [Google Scholar]
- 11.Freudenheim JL, Graham S, Marshall JR, et al. Folate intake and carcinogenesis of colon and rectum. Int J Epidemiol 1991;20:368–74. [DOI] [PubMed] [Google Scholar]
- 12.Bird CL, Swendseid ME, Witte JS, et al. Red cell and plasma folate, folate consumption and risk of colorectal adenomatous polyps. Cancer Epidemiol Biomarkers Prev 1995;4:709–14. [PubMed] [Google Scholar]
- 13.Paspatis G, Xourgias B, Mylonakou E, et al. A prospective clinical trial to determine the influence of folate supplementation on the formation of recurrent colonic adenomas. Gastroenterology 1994;106:A425. [Google Scholar]
- 14.Tseng M, Murray SC, Kupper LL, et al. Micronutrients and risk of colorectal adenomas. Am J Epidemiology 1995;141:S71. [DOI] [PubMed] [Google Scholar]
- 15.Giovannucci E, Stampfer MJ, Colditz GA, et al. Folate, methionine and alcohol intake and risk of colorectal adenoma. J Natl Cancer Inst 1993;85:875–84. [DOI] [PubMed] [Google Scholar]
- 16.Baron JA, Sandler RS, Haile RW, et al. Folate intake, alcohol consumption, cigarette smoking, and risk of colorectal adenoma. J Natl Cancer Inst 1998;90:57–62. [DOI] [PubMed] [Google Scholar]
- 17.Cravo ML, Mason JB, Dayal Y, et al. Folate deficiency enhances the development of colonic neoplasia in dimethylhydrazine-treated rats. Cancer Res 1992;52:5002–6. [PubMed] [Google Scholar]
- 18.Kim YI, Choi SW, Salomon RN, et al. Dietary folate protects against the development of macroscpoic colonic neoplasms in a dose responsive manner in the dimethylhydrazine rat model. Gastrointestinal Oncol 1995;108:A489. [Google Scholar]
- 19.Lipkin M. Biomarkers of increased susceptibility to gastrointestinal cancer: new application to studies of cancer prevention in human subjects. Cancer Res 1988;48:235–45. [PubMed] [Google Scholar]
- 20.Wilson RH, Williamson K, Gardiner TA, et al. Cell proliferation as a risk marker for colorectal cancer. J Irish Coll Phys Surg 1993;22:226–7. [Google Scholar]
- 21.Roncucci L, Scalmati A, Ponz de Leon M. Pattern of cell kinetics in colorectal mucosa of patients with different types of adenomatous polyps of the large bowel. Cancer 1991;68:873–8. [DOI] [PubMed] [Google Scholar]
- 22.Ponz de Leon M, Roncucci L, Di Donato P, et al. Pattern of epithelial cell proliferation in colorectal mucosa of normal subjects and of patients with adenomatous polyps or cancer of the large bowel. Cancer Res 1988;48:4121–6. [PubMed] [Google Scholar]
- 23.O'Sullivan KR, Mathias PM, Beattie S, et al. The effect of colonic site of biopsy on cell proliferation profiles using BRDU incorporation as a measure of cell proliferation. Eur J Cancer Prev 1992;1:381–3. [DOI] [PubMed] [Google Scholar]
- 24.Terpstra OT, van Blankenstein M, Dees J, et al. Abnormal pattern of cell proliferation in the entire colonic mucosa of patients with colon adenoma or cancer. Gastroenterology 1987;92:704–8. [DOI] [PubMed] [Google Scholar]
- 25.Hall C, Youngs D, Keighley MRB. Crypt cell production rates at various sites around the colon in Wistar rats and humans. Gut 1992;33:1528–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dolecek TA, Stamler J, Caggiula AW, et al. Methods of dietary and nutritional assessment and intervention and other methods in the multiple risk factor intervention trial. Am J Clin Nutr 1997;65:196–210S. [DOI] [PubMed] [Google Scholar]
- 27.Lipkin M. Biomarkers of increased susceptibility to gastrointestinal cancer. Gastroenterology 1987;92:1083–6. [DOI] [PubMed] [Google Scholar]
- 28.Ma Q, Williamson KE, O'Rourke D, et al. The effect of l-arginine on crypt cell hyperproliferation in colorectal cancer. J Surg Res 1999;81:181–8. [DOI] [PubMed] [Google Scholar]
- 29.Wargovich MJ, Alnutt D, Palmer C, et al. Inhibition of the promotional phase of azoxymethane-induced colon carcinogenesis in the F344 rat by calcium lactate: Effect of stimulating two different nutrient density levels. Cancer Lett 1990;53:17. [DOI] [PubMed] [Google Scholar]
- 30.Biasco G, Zannoni U, Paganelli GM, et al. Folic acid supplementation and cell kinetics of rectal mucosa in patients with ulcerative colitis. Cancer Epidemiol Biomarkers Prev 1997;6:469–71. [PubMed] [Google Scholar]
- 31.Department of Health (2000). Folic acid and the prevention of disease. Report on Health and Social Subjects 50. London: The Stationery Office, 2000.
- 32.Feinberg AP, Gehrke CW, Kuo KC, et al. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 1988;48:1159–61. [PubMed] [Google Scholar]
- 33.Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal tumour development. N Engl J Med 1988;319:525–32. [DOI] [PubMed] [Google Scholar]
- 34.Cravo M, Fidalgo P, Pereira AD, et al. DNA methylation as an intermediate biomarker of colorectal cancer: modulation by folic acid supplementation. Eur J Cancer Prev 1994;3:473–9. [DOI] [PubMed] [Google Scholar]
- 35.Kim YI, Pogribny IP, Basnakien AG, et al. Folate deficiency in rats induces DNA strand breaks and hypomethylation within the p53 tumour suppressor gene. Am J Clin Nutr 1997;65:46–52. [DOI] [PubMed] [Google Scholar]
- 36.Sedwick WD, Laszlo J. An antifolate-induced lesion in newly synthesized DNA. Adv Enzyme Regul 1980;19:295–308. [DOI] [PubMed] [Google Scholar]
- 37.Choi SW, Kim YL, Weitzel JN, et al. Folate depletion impairs DNA excision repair in the colon of the rat. Gut 1998;43:93–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mason JB, Levesque T. Folate: effects on carcinogenesis and the potential for cancer chemoprevention. Oncology 1996;10:1729–36. [PubMed] [Google Scholar]
- 39.Cho KR, Fearon ER. DCC: Linking tumour suppressor genes and altered cell surface interactions in cancer. Curr Opin Genet Dev 1995;5:72–8. [DOI] [PubMed] [Google Scholar]
- 40.Mason JB. Folate and colonic carcinogenesis: searching for a mechanistic understanding. J Nutr Biochem 1994;5:170–5. [Google Scholar]