

Abstract
Background: A homozygous mutation of the endothelin B receptor (EDNRB) gene in spotting lethal (sl/sl) rats leads to Hirschsprung's disease (HSCR) with long segmented aganglionosis. However, the effects on the development of the enteric nervous system (ENS) promoted by a heterozygous mutation of the EDNRB gene are not known. The present study aimed to describe and morphometrically assess the phenotypic abnormalities of the ENS in heterozygous (+/sl) EDNRB deficient rats in comparison with homozygous (sl/sl) EDNRB deficient and wild-type (+/+) rats.
Methods: The distal small intestine, caecum, and colon were obtained from sl/sl, +/sl, and +/+ rats. To demonstrate the three dimensional organisation of the ENS, the intestinal wall was microdissected into wholemounts and incubated against the pan-neuronal marker protein gene product 9.5. Assessment of the ENS included morphometric quantification of ganglionic size and density, the number of nerve cells per ganglia, and the diameter of nerve fibre strands within both the myenteric and submucous plexus.
Results: Sl/sl rats were characterised by complete aganglionosis resembling the same histopathological features observed in patients with HSCR. +/sl rats revealed more subtle abnormalities of the ENS: the submucous plexus was characterised by a significantly increased ganglionic size and density, and the presence of hypertrophied nerve fibre strands. Morphometric evaluation of the myenteric plexus did not show statistically significant differences between +/sl and +/+ rats.
Conclusions: In contrast with sl/sl rats, +/sl rats display non-aganglionated malformations of the ENS. Interestingly, these innervational abnormalities resemble the histopathological criteria for intestinal neuronal dysplasia (IND). Although IND has been described in several intestinal motility disorders, the concept of a clearly defined clinical-histopathological entity is still controversially discussed. The present findings support the concept of IND based on clearly defined morphological criteria suggesting a genetic link, and thus may provide a model for human IND. Furthermore, the data underline the critical role of the “gene dose” for the phenotypic effects promoted by the EDNRB/EDN3 system and confirm that the development of the ENS is not an “all or none” phenomenon.
Keywords: endothelin B receptor, enteric nervous system, Hirschsprung's disease, intestinal neuronal dysplasia, spotting lethal rat
Hirschsprung's disease (HSCR) is a congenital malformation of the enteric nervous system (ENS), regarded as a multigenic neurocristopathy,1 and represents a cause of significant paediatric morbidity and mortality.2 The characteristic histopathological features of HSCR are complete absence of ganglia in both the myenteric and submucous plexus and hypertrophy of nerve fibre strands within distal segments of the gastrointestinal tract. In the last decade different gene mutations have been identified in patients with HSCR.3
The term intestinal neuronal dysplasia (IND) was first introduced by Meier-Ruge in 19714 to describe hyperplastic changes of the submucous plexus underlying intestinal motility disorders with symptoms similar to those observed in HSCR. IND has been reported in adults and children suffering from chronic constipation and is frequently associated with HSCR. Giant ganglia and hypertrophied nerve fibre strands in the submucous plexus are the most prominent histopathological features of IND.5,6 However, the aetiology of IND is not yet known and the concept of a clearly defined clinical-histopathological entity is still controversially discussed.7 As HSCR and IND frequently occur in combination,8 similar molecular defects may underlie these distinct developmental disorders of the ENS.
Ontogenetic studies revealed that mutations in the endothelin B receptor (EDNRB) gene or its specific ligand endothelin-3 (EDN3) lead to defects in the development of neural crest cells.9,10 When colonisation of the gut by neural crest cells is incomplete, the distal part of the intestine is left aganglionic. A functional EDNRB/EDN3 system normally prevents the premature differentiation of crest derived precursor cells enabling the precursor population to persist long enough to finish colonisation of the fetal bowel.11,12 Thus one of the genes involved in the pathogenesis of HSCR is EDNRB.13 In humans, mutations in the EDNRB and EDN3 gene13,14 are probably carried by 10% of patients with HSCR.15 To date, at least 13 different mutations of the EDNRB gene associated with intestinal aganglionosis have been identified.16–18 A homozygous EDNRB deficiency leads to Waardenburg syndrome type IV (Shah-Waardenburg syndrome), a combination of long segmented HSCR and Waardenburg syndrome II characterised by sensorineural hearing loss and a pigmentary disorder.14,19,20 This phenotype is not only observed in humans but in all animal models in which the EDNRB or EDN3 gene is homozygously disrupted or mutated.21,22
The spotting lethal (sl) rat with homozygous EDNRB deficiency, an established animal model for HSCR,23,24 develops megaintestines caused by a long segmented aganglionosis resembling the same histopathological features observed in human HSCR.24 Homozygous (sl/sl) rats are characterised by white coat colour with pigmented spots25 and show a 301 bp deletion in both EDNRB genes.23 Rats carrying this deletion in only one of the EDNRB genes (heterozygous EDNRB deficient rats, +/sl) do not develop HSCR and pigmental abnormalities, but minor abnormalities of the myenteric plexus have been suggested.26 In humans, heterozygous EDNRB gene mutations also do not show an associated phenotype of Waardenburg type II but may cause HSCR. However, the penetrance of the HSCR phenotype is incomplete—in homozygous mutations (74%) less than in heterozygous (21%). Malformations of the ENS in heterozygous and homozygous individuals without HSCR are not known.3
Therefore, the aim of the present study was to assess possible abnormalities of the ENS in +/sl rats with a 301 bp deletion of one EDNRB gene. Whether a heterozygous mutation of the EDNRB gene is capable of provoking malformations of the ENS resembling the histopathological features of IND has yet to be verified.
MATERIALS AND METHODS
Rats
Animals from the Wistar-Imamichi AR strain (congenital aganglionosis) were used as homozygous (sl/sl) rats.23,27 They were bred by mating of heterozygous (+/sl) rats. The rats have an autosomal recessive 301 bp deletion in the EDNRB gene spanning exon 1 and intron 1, corresponding to the first and second transmembrane domains of the EDNRB.25 Animals were weaned from the mother at 21 days of age and maintained on standard rodent chow ad libitum. The genotype of each rat used for experiments was confirmed by polymerase chain reaction using primers flanking the 301 bp deletion of the mutant EDNRB gene.28 Four week old +/sl rats (n=5) were compared with two week old sl/sl (n=5) and four week old wild-type (+/+) rats (n=5).
Intestinal wholemounts and immunohistochemistry
After deep ether anaesthesia, rats were sacrificed and the intestines were removed immediately. The small intestine, caecum, and colon were filled with a fixation solution (3% phosphate buffered paraformaldehyde, 0.2% picric acid) for two hours. To perform a morphometric assessment, unbiased by the degree of intestinal distension, specimens obtained from +/+ and +/sl rats were equally stretched resulting in the same luminal diameters of the small intestine, caecum, and colon, respectively. After fixation, the intestinal segments were split open along the mesenteric border and washed 4×10 minutes in 50% ethanol. To improve the conditions for immunohistochemistry, specimens were rinsed in 0.1 M sodium phosphate buffer and stored overnight at 4°C in the same solution supplemented with 0.05% thimerosal. After treatment with 0.1% NaCNBH3 for 30 minutes, specimens were washed and stored at 4°C in 0.1 M sodium phosphate containing 0.01% NaN3.
Intestinal wholemounts of both the tunica muscularis and submucosa were dissected under stereomicroscopic control. After pretreatment with 10% normal goat serum (Dakopatts, Copenhagen, Denmark; X 907) for 30 minutes they were incubated in an antiserum raised against protein gene product 9.5 (Ultraclone RA 95101, diluted 1:400) for 24 hours.29,30 Goat antirabbit IgG (Dakopatts Z 421, diluted 1:100) was used as secondary antiserum. The wholemounts were then incubated in a solution containing a peroxidase antiperoxidase complex (Dakopatts Z 113, diluted 1:100) for 12 hours. 4-Chloro-1-naphthol was used as chromogen for the peroxidase reaction.
Morphometry and statistical analysis
In addition to the descriptive histopathological assessment of innervational abnormalities, the ENS of +/sl and +/+ rats was subjected to morphometric analysis. Measurements included registration of ganglia, nerve cells, and nerve fibre strands, and were carried out separately for the myenteric and submucous plexus. Evaluation was performed in a blinded fashion by two raters. For all of the measurements, interrater reliability ranged from 0.88 to 0.96 (p<0.0001).
The intestine of every animal was dissected into 24 wholemounts, 12 of the tunica muscularis and 12 of the submucosa, corresponding to the small intestine (n=10), caecum (n=4), and colon (n=10). In each wholemount the diameter of 10 randomly selected nerve fibre strands was recorded and a mean value calculated. Mean ganglionic density was calculated by counting the number of ganglia in four different areas (1 mm2) within a given wholemount. To determine mean ganglionic size, the area of eight ganglia was measured using the manual segmentation function of the software package Kontron KS 100 (Zeiss, Germany) which allows manual tracing of the regions of interest. Neuronal content per ganglion was determined by counting the number of nerve cells located within 10 ganglia.
Data were pooled for each group and evaluated separately for the intestinal segments examined (small intestine, caecum, colon). Statistical comparison between +/+ rats and +/sl rats was carried out by non-parametric two-tailed Mann-Whitney U tests with p≤0.05 considered as an indicator of significance.
RESULTS
Macroscopic findings
The colon, caecum, and distal ileum of sl/sl rats (fig 1A ▶) were constricted causing a long segmented “megaileum” (fig 1D ▶). The intestines of both +/sl (fig 1B ▶) and +/+ (fig 1C ▶) rats showed normal macroscopic features (fig 1E, 1F ▶). Only in one animal in the +/sl group was the caecum reduced in size (fig 1E ▶). Nevertheless, no +/sl rat showed clinical signs of functional intestinal obstruction and all developed normally similar to wild-type rats. Sl/sl rats died within the first three postnatal weeks.
Figure 1.
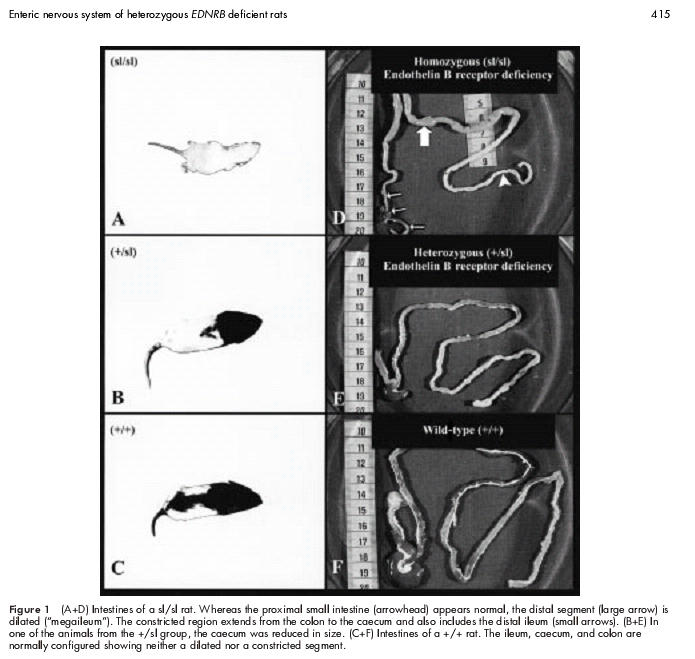
(A+D) Intestines of a sl/sl rat. Whereas the proximal small intestine (arrowhead) appears normal, the distal segment (large arrow) is dilated (“megaileum”). The constricted region extends from the colon to the caecum and also includes the distal ileum (small arrows). (B+E) In one of the animals from the +/sl group, the caecum was reduced in size. (C+F) Intestines of a +/+ rat. The ileum, caecum, and colon are normally configured showing neither a dilated nor a constricted segment.
Histopathological findings
Within the constricted intestinal segments of sl/sl rats, ganglia of both the myenteric and submucous plexus were absent (figs 2A, 3A ▶ ▶). Whereas the myenteric plexus was characterised by thickened nerve fibre strands running in a caudocranial direction, the submucous plexus was composed of a dense nerve network of irregularly distributed nerve fibre strands. The hypertrophied nerve fibre strands extended orally towards the transition zone. Within the dilated intestinal segment the nerve fibre strands became thinner and ramified into smaller branches. Proximal to the megaileum the myenteric and submucous plexus were normally configured showing the characteristic features of a regular ganglionic nerve network.
Figure 2.
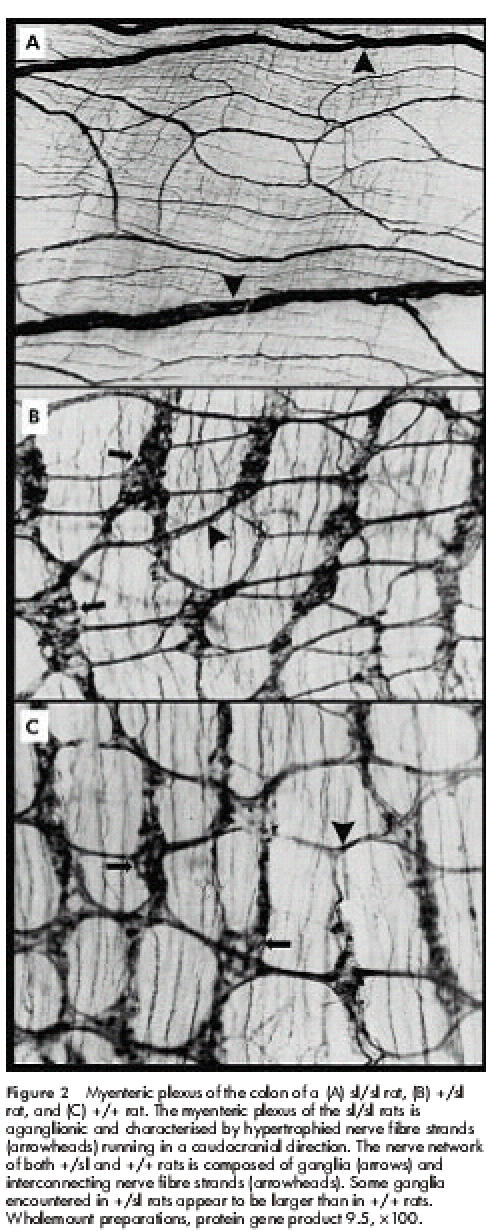
Myenteric plexus of the colon of a (A) sl/sl rat, (B) +/sl rat, and (C) +/+ rat. The myenteric plexus of the sl/sl rats is aganglionic and characterised by hypertrophied nerve fibre strands (arrowheads) running in a caudocranial direction. The nerve network of both +/sl and +/+ rats is composed of ganglia (arrows) and interconnecting nerve fibre strands (arrowheads). Some ganglia encountered in +/sl rats appear to be larger than in +/+ rats. Wholemount preparations, protein gene product 9.5, ×100.
Figure 3.
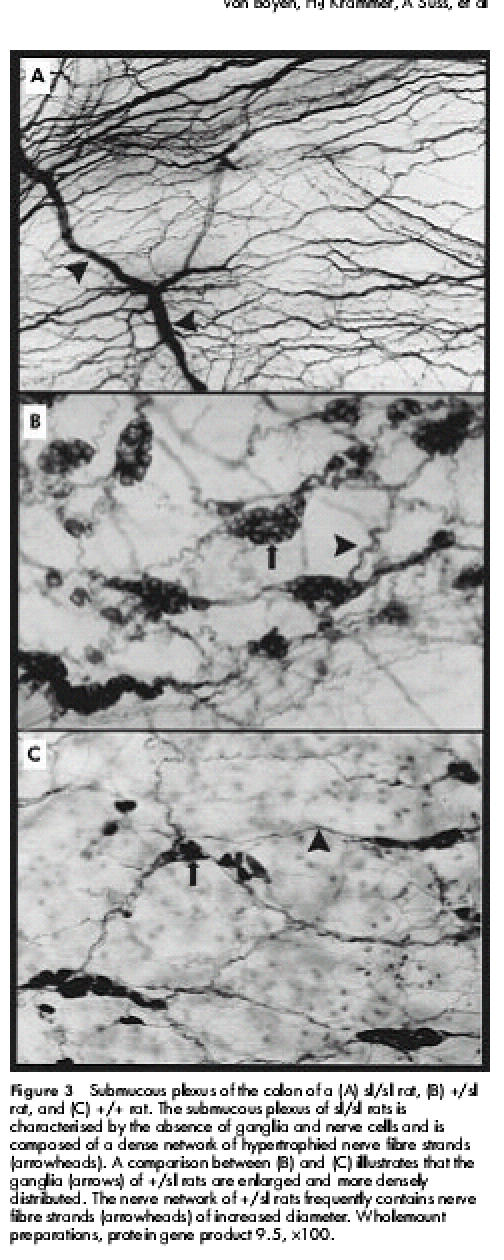
Submucous plexus of the colon of a (A) sl/sl rat, (B) +/sl rat, and (C) +/+ rat. The submucous plexus of sl/sl rats is characterised by the absence of ganglia and nerve cells and is composed of a dense network of hypertrophied nerve fibre strands (arrowheads). A comparison between (B) and (C) illustrates that the ganglia (arrows) of +/sl rats are enlarged and more densely distributed. The nerve network of +/sl rats frequently contains nerve fibre strands (arrowheads) of increased diameter. Wholemount preparations, protein gene product 9.5, ×100.
+/sl rats did not show distal aganglionosis, as observed in sl/sl rats. However, in comparison with +/+ animals, +/sl rats revealed the following structural abnormalities: the myenteric plexus frequently contained ganglia of larger size and the primary nerve fibre strands appeared to be slightly thickened (fig 2B, 2C ▶ ▶). These findings were even more pronounced within the submucous nerve network which exhibited an obvious increase in ganglionic density and size (fig 3B, 3C ▶). Frequently the borders of adjacent ganglia fused to form “ganglionic conglomerates”. Higher magnification showed that the ganglia of +/sl rats contained an increased number of neurones compared with +/+ rats (fig 4A, 4B ▶). Moreover, the diameter of submucous nerve fibre strands appeared to be enlarged, in particular within the caecum and colon of +/sl rats. However, no nerve cell degeneration or signs of neuronal immaturity (neurones of reduced size and poorly developed cytoplasm) were observed.
Figure 4.
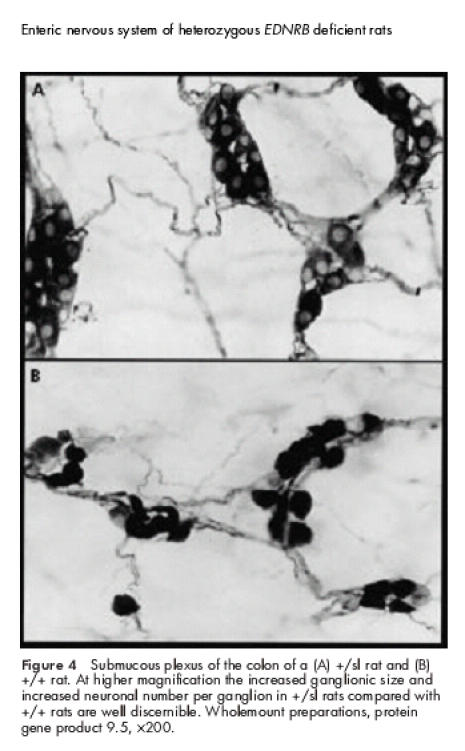
Submucous plexus of the colon of a (A) +/sl rat and (B) +/+ rat. At higher magnification the increased ganglionic size and increased neuronal number per ganglion in +/sl rats compared with +/+ rats are well discernible. Wholemount preparations, protein gene product 9.5, ×200.
Morphometric analysis
Whereas the aganglionic condition in sl/sl rats was readily discernible, the less severe abnormalities of the ENS encountered in +/sl rats, as described above, were subjected to morphometric analysis to verify if the alterations differed significantly from +/+ rats (tables 1–3 ▶ ▶ ▶). With regard to the myenteric plexus, no statistically significant differences were found between +/+ and +/sl rats concerning ganglionic size and density, neuronal content, or diameter of nerve fibre strands. However, statistical comparison of the data obtained for the submucous plexus confirmed that the ganglia of +/sl rats were significantly larger, contained more neurones, and were more densely distributed. Mean ganglionic density of +/sl rats exceeded that of +/+ rats by about two times in the small intestine, three times in the caecum, and more than five times in the colon. A similar craniocaudal gradient was also recorded for ganglionic size and neuronal content, indicating that the hyperplastic changes of the submucous plexus were more pronounced within the distal segments of the gut. This observation was also made with regard to the nerve fibre strands of the submucous plexus: whereas in the caecum and colon of +/sl rats the mean diameters differed significantly from +/+ rats (two times larger), in the small intestine no statistically significant differences were found.
Table 1.
Statistical comparison between +/+ rats and +/sl rats—small intestine
| +/+ rats | +/sl rats | p Value | |
| Ganglionic size (μm2) | |||
| Myenteric plexus | 7077 (883.8) | 7782 (248.4) | NS |
| Submucous plexus | 1643 (296.4) | 5127 (484.6) | <0.05 |
| Neuronal No per ganglion | |||
| Myenteric plexus | 13.87 (2.16) | 13.56 (2.32) | NS |
| Submucous plexus | 4.40 (1.48) | 10.20 (1.23) | <0.05 |
| Ganglionic density (No/mm2) | |||
| Myenteric plexus | 12.32 (1.27) | 12.79 (2.00) | NS |
| Submucous plexus | 9.68 (1.21) | 21.62 (1.28) | <0.05 |
| Diameter of nerve fibre strands (μm) | |||
| Myenteric plexus | 5.83 (1.34) | 6.98 (2.01) | NS |
| Submucous plexus | 4.81 (0.39) | 7.07 (2.32) | NS |
Data are mean (SD).
Table 2.
Statistical comparison between +/+ rats and +/sl rats—caecum
| +/+ rats | +/sl rats | p Value | |
| Ganglionic size (μm2) | |||
| Myenteric plexus | 7495 (919.2) | 9104 (122.5) | NS |
| Submucous plexus | 1979 (210.8) | 8143 (501.5) | <0.05 |
| Neuronal No per ganglion | |||
| Myenteric plexus | 16.63 (1.37) | 18.24 (0.93) | NS |
| Submucous plexus | 4.30 (1.27) | 13.33 (0.86) | <0.05 |
| Ganglionic density (No/mm2) | |||
| Myenteric plexus | 9.80 (0.95) | 11.53 (1.68) | NS |
| Submucous plexus | 6.92 (0.64) | 20.94 (0.94) | <0.05 |
| Diameter of nerve fibre strands (μm) | |||
| Myenteric plexus | 7.91 (1.64) | 9.83 (1.26) | NS |
| Submucous plexus | 5.48 (0.41) | 10.66 (1.47) | <0.05 |
Data are mean (SD).
Table 3.
Statistical comparison between +/+ rats and +/sl rats—colon
| +/+ rats | +/sl rats | p Value | |
| Ganglionic size (μm2) | |||
| Myenteric plexus | 8365 (784.6) | 9983 (302.8) | NS |
| Submucous plexus | 2212 (346.1) | 8277 (687.4) | <0.05 |
| Neuronal No per ganglion | |||
| Myenteric plexus | 15.76 (2.92) | 20.81 (2.62) | NS |
| Submucous plexus | 6.10 (0.9) | 13.92 (1.51) | <0.05 |
| Ganglionic density (No /mm2) | |||
| Myenteric plexus | 6.99 (1.22) | 10.01 (1.93) | NS |
| Submucous plexus | 3.14 (0.46) | 17.00 (1.18) | <0.05 |
| Diameter of nerve fibre strands (μm) | |||
| Myenteric plexus | 8.52 (1.53) | 9.77 (1.14) | NS |
| Submucous plexus | 5.32 (0.23) | 10.51 (2.11) | <0.05 |
Data are mean (SD).
DISCUSSION
In the present study, we showed that a heterozygous 301 bp deletion of the EDNRB gene leads to morphological abnormalities of the ENS. The statistically significant alterations primarily affected the submucous plexus and consisted of an increase in ganglionic size and density, increased neuronal content of the ganglia, and hypertrophy of nerve fibre strands. Obviously, a heterozygous defect does not completely abolish the inhibitory potential of the EDNRB/EDN3 system11 allowing the gut to be colonised by precursor cells. However, loss of one functional EDNRB gene may cause premature formation of “ganglionic conglomerates” before the arrangement of nerve cells into normal sized ganglia has been completed. As the submucosa is the last intestinal layer to be colonised during the intramural migration of precursor cells, incomplete development of the ENS will affect the architecture of the submucous nerve network in particular. Moreover, as the EDNRB/EDN3 system mainly influences neuronal development within distal segments of the gut,11,12 malformations of the ENS will be expected primarily in this region. Indeed, the morphometric analysis of the ENS from +/sl rats confirmed that the most pronounced abnormalities were located within the caecum and colon and only to a lesser degree within the small intestine.
Interestingly, malformations of the ENS observed in +/sl rats, such as submucous giant ganglia and hypertrophied nerve fibre strands, resemble the histopathological features of IND described in humans.5,6 IND has been reported as an enteric neuropathology underlying severe chronic constipation and intestinal pseudo-obstruction in children and adults.31–33 Controversies concerning the reliability of the diagnosis (for example, histopathological criteria, morphometric techniques) and clinical management (for example, conservative versus surgical therapy) have questioned IND as a clinical-histopathological entity.34,35 However, morphometric analysis of the ENS from +/sl rats carried out in the present study clearly revealed alterations similar to those found in IND—for example, hyperganglionosis of the submucous plexus and hypertrophy of nerve fibre strands. Thus at least from a morphological point of view, our findings support the concept of IND as a distinct intestinal innervational disorder. Hyperganglionosis of the enteric nerve plexus has also been described recently in a mouse model carrying a Ncx (Enx, Hox11L1) deficiency and was proposed as a model for human IND.36,37 These data suggest that the same phenotypic abnormality of the ENS may be caused by multiple genetic factors, as has been shown for HSCR.3
Although the pathophysiological significance of an increased number of neurones is not well established, it has been reported that neuronal hyperplasia in Ncx (Enx, Hox11L1) deficient mice leads to abnormal intestinal peristalsis37 and that an increased number of hippocampal neurones in fyn deficient mice causes learning abnormalities.38 Thus hyperinnervation of the submucous plexus may cause inappropriate activation of enteric neurones. As the submucous plexus is actively involved in the modulation of intestinal motility,39 alterations in this nerve network—apart from abnormalities of the myenteric plexus—may also provoke intestinal motor dysfunctions leading to symptoms of chronic constipation.
In the early postnatal period, +/sl rats showed no obvious signs of functional intestinal obstruction. We cannot exclude the fact that the enteric neuronal malformations may be clinically compensated over a given time period and may not lead to intestinal motility disorders until adulthood. Whereas +/sl rats did not show the histopathological and clinical features of HSCR, in about 5% of patients with HSCR a heterozygous EDNRB deficiency has been found.3 It is not known whether patients without HSCR, who carry a heterozygous EDNRB deficiency, show abnormalities of their ENS similar to those observed in +/sl rats. If the EDNRB gene mutation is heterozygous, less severe enteric neuronal malformations such as IND may be expected due to weaker expression of the gene product. Systematic genetic screening for a heterozygous mutation in the EDNRB gene in a representative group of patients definitely diagnosed with IND has not yet been carried out. However, recently it has been shown that in a small number of patients with IND (n=20) and combined HSCR/IND (n=12), an EDNRB mutation could not be identified.40 This is not surprising considering the frequency of EDNRB mutations in HSCR of only 2–4%.3 The same is also true for those HSCR patients who have been additionally diagnosed with IND. Moreover, the absence of a genetic defect in patients with IND can also be attributed to the fact that abnormalities of the ENS may arise not only from congenital but also from environmental factors. In particular, hyperplastic changes, as observed in IND, have also been described in experimental intestinal stenosis reflecting the postnatal plasticity of the ENS.41
In contrast with SOX10, endothelin converting enzyme, and other factors (for example, GDNF protein family, neurotrophins, neuropoietic cytokines),42–46 the EDNRB/EDN3 system acts as an inhibitory tool in enteric neuronal differentiation.11 Whereas a functional EDNRB/EDN3 system prevents differentiation of precursor cells before having colonised the entire bowel wall, a homozygous defect causes premature differentiation leaving the distal gut aganglionic. The present study provides morphometrically confirmed evidence that the phenotype of the ENS is not only altered by a homozygous but also by a heterozygous 301 bp deletion of the EDNRB gene: whereas the homozygous defect causes long segmented HSCR, heterozygous EDNRB deficiency leads to an IND-like histopathology. The latter observation suggests that hyperganglionosis may be linked to a genetic defect and that +/sl rats may provide another model for human IND. Furthermore, the data underline the critical role of the “gene dose” for the phenotypic effects promoted by the EDNRB/EDN3 system and confirm that the development of the ENS is not an “all or none” phenomenon.
Abbreviations
HSCR, Hirschsprung's disease
ENS, enteric nervous system
IND, intestinal neuronal dysplasia
EDNRB, endothelin B receptor
EDN3, endothelin-3
sl rat, spotting lethal rat
sl/sl, homozygous
+/sl, heterozygous
+/+, wild-type
REFERENCES
- 1.Garver KL, Garner B, Law JC. Hirschsprung's disease: a genetic study. Clin Genet 1985;26:261–77. [DOI] [PubMed] [Google Scholar]
- 2.Skinner MA. Hirschsprung's disease. Curr Probl Surg 1996;33:389–460. [DOI] [PubMed] [Google Scholar]
- 3.Kusafuka T, Puri P. Genetic aspects of Hirschsprung's disease. Semin Pediatr Surg 1998;7:148–55. [DOI] [PubMed] [Google Scholar]
- 4.Meier-Ruge W. Causes of colon disorder with symptoms of Hirschsprung's disease. Verh Dtsch Ges Pathol 1971;55:506–10. [PubMed] [Google Scholar]
- 5.Borchard F, Meier-Ruge W, Wiebecke B, et al. (Disorders of the innervation of the large intestine-classification and diagnosis. Results of a consensus conference of the Society of Gastroenteropathology, 1 December 1990 in Frankfurt/Main). Pathologe 1991;12:171–4. [PubMed] [Google Scholar]
- 6.Meier-Ruge WA, Bronnimann PB, Gambazzi F, et al. Histopathological criteria for intestinal neuronal dysplasia of the submucosal plexus. Virchows Arch 1995;426:549–56. [DOI] [PubMed] [Google Scholar]
- 7.Berry CL. Intestinal neuronal dysplasia: does it exist or has it been invented? Virchows Arch A Pathol Anat Histopathol 1993;422:183–4. [DOI] [PubMed] [Google Scholar]
- 8.Holschneider AM, Meier-Ruge W, Ure BM. Hirschsprung's disease and allied disorders—a review. Eur J Pediatr Surg 1994;4:260–6. [DOI] [PubMed] [Google Scholar]
- 9.Greenstein Baynash A, Hosoda K, Giaid A, et al. Interaction of endothelin-3 with endothelin-B receptor is essential for development of epidermal melanocytes and enteric neurons. Cell 1994;79:1277–85. [DOI] [PubMed] [Google Scholar]
- 10.Hosoda K, Hammer RE, Richardson JA, et al. Targeted and natural (piebald-lethal) mutations of endothelin-B-receptor gene produce megacolon associated with spotted coat color in mice. Cell 1994;79:1267–76. [DOI] [PubMed] [Google Scholar]
- 11.Wu JJ, Chen JX, Rothman TP, et al. Inhibition of in vitro enteric neuronal development by endothelin-3: mediation by endothelin B receptors. Development 1999;126:1161–73. [DOI] [PubMed] [Google Scholar]
- 12.Gershon MD. Endothelin and the development of the enteric nervous system. Clin Exp Pharmacol Physiol 1999;26:985–88. [DOI] [PubMed] [Google Scholar]
- 13.Puffenberger EG, Hosoda K, Washington SS, et al. A missense mutation of the endothelin-B receptor gene in multigenic Hirschsprung's disease. Cell 1994;79:1257–66. [DOI] [PubMed] [Google Scholar]
- 14.Hofstra RM, Osinga J, Tan-Sindhunata G, et al. A homozygous mutation in the endothelin-3 gene associated with a combined Waardenburg type 2 and Hirschsprung phenotype (Shah-Waardenburg syndrome). Nat Genet 1996;12:445–7. [DOI] [PubMed] [Google Scholar]
- 15.Bidaud C, Salomon R, Van Camp G, et al. Endothelin-3 gene mutations in isolated and syndromic Hirschsprung disease. Eur J Hum Genet 1997;5:247–51. [PubMed] [Google Scholar]
- 16.Attie T, Till M, Pelet A, et al. Mutation of the endothelin-receptor B gene in Waardenburg-Hirschsprung's disease. Hum Mol Genet 1995;4:2407–9. [DOI] [PubMed] [Google Scholar]
- 17.Auricchio A, Casari G, Staiano A, et al. Endothelin-B-receptor mutations in patients with isolated Hirschsprung's disease from a noninbred population. Hum Mol Genet 1996;5:351–4. [DOI] [PubMed] [Google Scholar]
- 18.Amiel J, Attie T, Jan D, et al. Heterozygous endothelin receptor B (EDNRB) mutations in isolated Hirschsprung disease. Hum Mol Genet 1996;5:355–7. [DOI] [PubMed] [Google Scholar]
- 19.Edery P, Attie T, Amiel J, et al. Mutation of the endothelin-3 gene in the Waardenburg-Hirschsprung disease (Shah-Waardenburg syndrome). Nat Genet 1996;12:442–4. [DOI] [PubMed] [Google Scholar]
- 20.Read AP, Newton VE. Waardenburg syndrome. J Med Genet 1997;34:656–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santschi E, Purdy A, Valberg S, et al. Endothelin receptor B polymorphism associated with lethal white foal syndrome in horses. Mamm Genome 1998;9:306–9. [DOI] [PubMed] [Google Scholar]
- 22.McCabe L, Griffin LD, Kinzer A, et al. Overo lethal white foal syndrome: equine model of aganglionic megacolon (Hirschsprung disease). Am J Med Genet 1990;36:336–40. [DOI] [PubMed] [Google Scholar]
- 23.Kunieda T, Kumagai T, Tsuji T, et al. A mutation in endothelin-B receptor gene causes myenteric aganglionosis and coat color spotting in rats. DNA Res 1996;3:101–5. [DOI] [PubMed] [Google Scholar]
- 24.Schneider (von Boyen) GBT, Wedel T, Krammer H-J, et al. The enteric nervous system in three dimensional intestinal wholemounts of homozygous endothelin-B-receptor (ETB) deficient (spotting lethal) rats compared to patients with Hirschsprung's disease. In: Krammer H-J, Singer MV, eds. Neurogastroenterology-from the basics to the clinics. London: Kluwer Academic Publishers, 2000:485–92.
- 25.Gariepy CE, Cass DT, Yanagisawa M. Null mutation of endothelin receptor type B gene in spotting lethal rats causes aganglionic megacolon and white coat color. Proc Natl Acad Sci USA 1996;93:867–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dembowski C, Hofmann P, Koch T, et al. Phenotype, intestinal morphology and survival of homozygous and heterozygous endothelin B receptor deficient (spotting lethal) rats. J Pediatr Surg 2000;35:480–8. [DOI] [PubMed] [Google Scholar]
- 27.Nagahama M, Ozaki T, Hama K. A study of the myenteric plexus of the congenital aganglionosis rat (spotting lethal). Anat Embryol 1985;171:285–96. [DOI] [PubMed] [Google Scholar]
- 28.Ehrenreich H, Oldenburg J, Hasselblatt M, et al. Endothelin B receptor deficient rats as a subtraction model to study the cerebral endothelin system. Neuroscience 1999;91:1067–75. [DOI] [PubMed] [Google Scholar]
- 29.Thompson RJ, Doran JF, Jackson P, et al. PGP 9.5—a new marker for vertebrate neurons and neuroendocrine cells. Brain Res 1983;278:224–8. [DOI] [PubMed] [Google Scholar]
- 30.Wilson POG, Barber PC, Hamid QA, et al. The immunolocalization of protein gene product 9.5 using rabbit polyclonal and mouse monoclonal antibodies. Br J Exp Pathol 1988;69:91–104. [PMC free article] [PubMed] [Google Scholar]
- 31.Wedel T, Roblick U, Gleiss J, et al. Disorders of intestinal innervation as a possible cause for chronic constipation. Zentralbl Chir 1999;124:796–803. [PubMed] [Google Scholar]
- 32.Schmittenbecher PP, Gluck M, Wiebecke B, et al. Clinical long-term follow-up results in intestinal neuronal dysplasia (IND). Eur J Pediatr Surg 2000;10:17–22. [DOI] [PubMed] [Google Scholar]
- 33.Voderholzer WA, Wiebecke B, Gerum M, et al. Dysplasia of the submucous nerve plexus in slow-transit constipation of adults. Eur J Gastroenterol Hepatol 2000;12:755–9. [DOI] [PubMed] [Google Scholar]
- 34.Puri P. Variant Hirschsprung's disease. J Pediatr Surg 1997;32:149–57. [DOI] [PubMed] [Google Scholar]
- 35.Koletzko S, Jesch I, Faus-Kebetaler T, et al. Rectal biopsy for diagnosis of intestinal neuronal dysplasia in children: a prospective multicentre study on interobserver variation and clinical outcome. Gut 1999;44:853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shirasawa S, Yunker AM, Roth KA, et al. Enx (Hox11L1)-deficient mice develop myenteric neuronal hyperplasia and megacolon. Nat Med 1997;3:646–50. [DOI] [PubMed] [Google Scholar]
- 37.Hatano M, Aoki T, Dezawa M, et al. A novel pathogenesis of megacolon in Ncx/Hox11L.1 deficient mice. J Clin Invest 1997;100:795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grant SG, O'Dell TJ, Karl KA, et al. Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 1992;258:1903–10. [DOI] [PubMed] [Google Scholar]
- 39.Kunze WAA, Furness JB. The enteric nervous system and regulation of intestinal motility. Annu Rev Physiol 1999;61:117–42. [DOI] [PubMed] [Google Scholar]
- 40.Gath R, Goessling A, Keller KM, et al. Analysis of the RET, GDNF, EDN3, and EDNRB genes in patients with intestinal neuronal dysplasia and Hirschsprung disease. Gut 2001;48:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gabella G. Size of neurons and glial cells in the intramural ganglia of the hypertrophic intestine of the guinea-pig. J Neurocytol 1984;13:73–84. [DOI] [PubMed] [Google Scholar]
- 42.Hearn CJ, Murphy M, Newgreen D. GDNF and ET-3 differentially modulate the numbers of avian enteric neural crest cells and enteric neurons in vitro. Dev Biol 1998;197:93–105. [DOI] [PubMed] [Google Scholar]
- 43.Moore MW, Klein RD, Farias I, et al. Renal and neuronal abnormalities in mice lacking GDNF. Nature 1996;382:76–9. [DOI] [PubMed] [Google Scholar]
- 44.Chalazonitis A, Rothman TP, Chen J, et al. Age-dependent differences in the effects of GDNF and NT-3 on the development of neurons and glia from neural crest-derived precursors immunoselected from the fetal rat gut: expression of GFR á in vitro and vivo. Dev Biol 1998;204:385–406. [DOI] [PubMed] [Google Scholar]
- 45.DeChiara TM, Vejsada R, Poueymirou WT, et al. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell 1995;83:313–22. [DOI] [PubMed] [Google Scholar]
- 46.Kapur RP. Hirschsprung's disease and other dysganglionoses. Crit Rev Clin Lab Sci 1999;36:225–73. [DOI] [PubMed] [Google Scholar]