

Abstract
Background: Cholangiocarcinoma cells express high levels of the antiapoptotic proteins Bcl-XL and Mcl-1 and are markedly chemo- and radioresistant. Mitochondria have emerged as central players in apoptosis. Antiapoptotic members of the Bcl-2 protein family localise to the outer mitochondrial membrane and regulate mitochondrial release of apoptogenic proteins. Mitochondrial benzodiazepine receptor (mBzR) ligands have been shown to reverse Bcl-2 action and facilitate apoptosis.
Aim: We evaluated the ability of the mBzR antagonist Pk11195 to overcome preapoptotic mitochondrial dysfunction in Egi-1 and Tfk-1, two human cholangiocarcinoma cell lines expressing high levels of Bcl-XL and Mcl-1.
Materials and methods: Cells growing in culture were used to perform in vitro experiments over 48–96 hours following treatment. The cytotoxic agents used were 5 fluorouracil 10 μM and etoposide (Vp16) 10 μM, together with ultraviolet and 0.5–1 Gy x ray irradiation with or without 75 μM Pk11195. Apoptosis and mitochondrial dysfunction were measured at single cell resolution by flow cytometry using the mitochondrial fluorochrome DiOC6(3). Severe combined immunodeficient non-obese diabetic (SCID-NOD) mice with subcutaneous xenografts using the Egi-1 and Tfk-1 cell lines were treated with etoposide with or without addition of Pk11195 over a 72 hour period during which time the xenograft growth patterns were monitored.
Results: In vitro, the effect of Pk11195 on induction of apoptosis in cholangiocarcinoma cells following stimulation by chemotherapy or radiotherapy was found to be both time and dose dependent, with Pk11195 increasing rates of apoptosis by 50–95%. Intraperitoneal administration of Pk11195 in combination with Vp16 was found to increase the growth inhibiting effects of Vp16 on xenografts during the treatment phase. PK11195 75 μM on its own had no intrinsic cytotoxic efficacy.
Conclusion: This is the first study to demonstrate that functional antagonism of coexpressed Bcl-XL and Mcl-1 proteins using the mBzR antagonist Pk11195 can facilitate apoptosis in cholangiocarcinoma following chemotherapy and radiotherapy.
Keywords: cholangiocarcinoma, mitochondria, apoptosis resistance, peripheral benzodiazepine receptor, Bcl-XL, Mcl-1
Cholangiocarcinoma carries a poor prognosis. Conventional anticancer treatments such as chemotherapy and radiotherapy have had minimal impact on patient survival in unresectable disease.1–6 The factors responsible for the relative unresponsiveness of cholangiocarcinoma to cytotoxic therapies have not been identified. The efficacy of conventional anticancer therapies is dependent on an ability to initiate programmed cell death (apoptosis) in cancer cells.7,8 Consequently, cancer cells that have evolved ways of avoiding apoptosis are rendered resistant, providing an obstacle to effective treatment.9 Apoptosis is a biochemically stereotyped, well conserved, and highly regulated mechanism essential for normal embryogenesis and maintenance of tissue homeostasis.10 The Bcl-2 family of apoptosis regulating proteins function either to promote (Bax, Bad, Bak) or inhibit (Bcl-2, Bcl-XL, and Mcl-1) the apoptotic response to a wide variety of stimuli, including chemotherapy and radiotherapy.11–18 In a recent study examining expression of Bcl-2, Mcl-1, and Bcl-XL in human cholangiocarcinoma, all 51 cases were found to coexpress Bcl-XL and Mcl-1 protein but not Bcl-2.19 Others have studied expression of Bcl-2 in cholangiocarcinoma with some groups detecting and others not detecting Bcl-2.20–22 Whether expression of antiapoptotic proteins influences the response of cholangiocarcinoma cells to chemotherapy or radiotherapy has not been investigated.
Pk11195 is a ligand of the mitochondrial benzodiazepine receptor (mBzR) which interacts with a common downstream target of Bcl-2 and Bcl-XL known as the permeability transition pore complex (PTPC). Pk11195 has been shown to antagonise Bcl-2 function23 by facilitating pore opening, induced by cytotoxic stimuli, leading to cessation of oxidative phosphorylation24 and activation of the caspase cascade that mediates the features of apoptosis. Resistance to apoptosis may explain the poor response of cholangiocarcinoma to chemotherapy and radiotherapy. This study has investigated the effect of the mBzR antagonist Pk11195 on mitochondrial dysfunction induced by a range of apoptosis inducing stimuli in Bcl-XL and Mcl-1 expressing human cholangiocarcinoma cell lines.
MATERIALS AND METHODS
In this study, Bcl-XL and Mcl-1 expressing human cholangiocarcinoma cells lines Egi-1 and Tfk-1 were treated in culture by chemotherapy, ultraviolet (UV), and x ray irradiation with or without addition of the mBzR antagonist Pk11195. Cellular apoptosis was measured. The same cell line was then implanted subcutaneously as xenografts on the back of severe combined immunodeficient non-obese diabetic (SCID-NOD) mice. The growth response of the xenografts to etoposide treatment was evaluated with or without addition of Pk11195.
Materials
Bcl-2 and the mBzR antagonist Pk11195, propidium iodide, and 5 fluorouracil were obtained from Sigma-Aldrich limited (Dorset, UK). The green coloured lipophilic mitochondrial probe dihexyloxalocardocyanine (DiOC6(3)) was obtained from Molecular Probes (Cambridge Bioscience, Cambridge, UK). Etoposide was obtained from Vepesid (Bristol Myers, UK). Mcl-1 and Bcl-XL rabbit antihuman polyclonal antibodies and intrastain fixation/permeabilisation kit were obtained from Dako Ltd (Ely, UK). The mBzR specific probe NBD FGIN 1–27 analogue was obtained from Alexis Biochemicals (Cambridge, UK) and annexin V fluorescein isothiocynate (FITC) was obtained from Bender Medsystems (Paris, France).
Cell lines
Egi-1 and Tfk-1 are two well characterised adherent human cholangiocarcinoma cell lines25 derived from patient cells prior to any exposure to chemotherapy or radiotherapy. Both cell lines are P-glycoprotein negative and express mitochondrial Bcl-XL and Mcl-1. Tfk-1 was cultured at 37°C with 5% CO2 in RPMI 1640 medium (Sigma) supplemented with 5 mM glutamine, 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Egi-1 was grown in 1:1 minimum essential medium (Sigma) and Dulbecco’s modified essential medium (Sigma) supplemented with 1 mM non-essential amino acids, 2 mM essential amino acids, 5 mM glutamine, penicillin-streptomycin supplements, and 10% FCS. Culture medium was replenished to avoid nutrient exhaustion every 48 hours during the experiments.
Assessment of antiapoptotic proteins in cell lines
Tfk-1 and Egi-1 were trypsinised 60–75% preconfluence using trypsin/EDTA (×1; Sigma). After washing in buffered saline, cells were fixed and permeabilised using the Dako intrastain kit. FITC conjugated rabbit polyclonal antihuman Mcl-1 and Bcl-XL antibodies and negative control rabbit immunoglobulin were incubated at 1 in 100 dilution in the dark at room temperature for 15 minutes. Cells were analysed at single cell resolution by flow cytometry (Becton Dickinson, Oxford, UK) with the aid of Cellquest software (version 3.2.1) for quantification of fluorescence intensity.
Detection of benzodiazepine receptor (mBzR) expression
The presence of the mBzR in cholangiocarcinoma cells was investigated using a specific mBzR fluorescent probe 7-nitro-2, 1, 3-benzoxadiazol-4-yl derivative (NBD FGIN-1–27 analogue)26 and mitochondrial fluorochrome chloromethyl-X-rosomine (CMXRos). Live cells suspended in culture medium were incubated for 45 minutes with 1 μM NBD FGIN-1–27 and CMXRos at 37°C. After being mounted on slides, images were captured using a Zeiss Axioskop fluorescence microscope running Iplab spectrum image analysis software (version 3.1.1).
Chemotherapy induced apoptosis
Flasks (75 cm2) containing exponentially growing cells that were approximately 60–70% confluent were split using trypsin/EDTA (×1). Cells (1×104) were then recultured in 12 well flat bottomed plates, allowed to adhere overnight, and treated the following day with either 10 μM 5 fluorouracil or 10 μM Vp16, with or without 75 μM Pk11195.
Ultraviolet and x ray irradiation induced apoptosis
Twelve well flat bottomed plates containing growing cells in culture were exposed to either UV irradiation (120 mJ/cm2; Chromato-UV-E transilluminator, model TM-20) for five minutes or x ray irradiation at a dose of 0.5–1 Gy from a 6 MV linear accelerator (Elekta oncology SL75/5), with or without Pk11195 at a dose of 75 μM
Flow cytometry
Multiparametric cell analysis was performed at single cell resolution by flow cytometry running the Cellquest software (version 3.2.1). At the appropriate time the experiment was terminated and all floating and adherent cells (after trysinisation) from each well were collected in well labelled FACS tubes, washed and spun down to a pellet, and then resuspended in culture medium ready for staining.
Measurement of mitochondrial permeability transition
Mitochondrial dysfunction, one of the parameters for detecting apoptosis, was measured at single cell level by detecting collapse of the inner mitochondrial membrane potential, ΔΨm.26–29 Cells resuspended in culture medium for analysis were incubated for 15 minutes in the dark at room temperature with the potentiometric probe 75 nM DiOC6(3) and 20 μg/ml propidium iodide. ΔΨm was measured by flow cytometry along the FL1 and FL3 channels acquiring 10 000 events (cells) per well. All data collected were analysed using software package WinMDI 2.8 windows multiple document interface for flow cytometry (TSRI, La Jolla, California, USA) and exported into a Microsoft excel sheet for statistics and chart generation.
Phosphotidylserine expression
This caspase dependent event was measured using the annexin V assay. Cells in suspension were incubated in calcium buffer and 5 μl FITC annexin V for 15 minutes in the dark. Cells were washed twice in Hank’s buffered saline solution and then resuspended in 20 μg/ml propidium iodide for 10 minutes before analysis by flow cytometry.
Measurement of antitumour activity in immunodeficient mice-xenograft models
SCID-NOD mice were allocated for interscapular subcutaneous inoculation of 5×107 Egi-1 or Tfk-1 cells in 1 ml of medium. Tumours grew in all cases and usually became palpable within 2–3 weeks. Using calipers, tumour size was measured along three different planes (diameters) and growth monitored. Mice were considered eligible for treatment when the normalised mean diameter of the tumour xenograft was 120–150% of its original size (first time measured, day 1 on graph), allowing a within xenograft comparison of pretreatment and post-treatment growth rates. Mice were randomly subdivided into three treatment groups (85 μg/mouse/day of Vp16, 85 μg/mouse/day of Vp16+10 mg/kg/day of Pk11195, and saline (control)) making a total of nine mice per single experiment (triplicates). Mice were treated daily for three days on two separate occasions (schedules 1 and 2) 16 days apart by daily intraperitoneal administration of Vp16+/−Pk11195 or saline. Tumour growth (Tt) was calculated using the mean of three tumour diameters (d1 to d3) at time t, and normalising against the initial tumour size measured at time t = 0 (T0), that is,
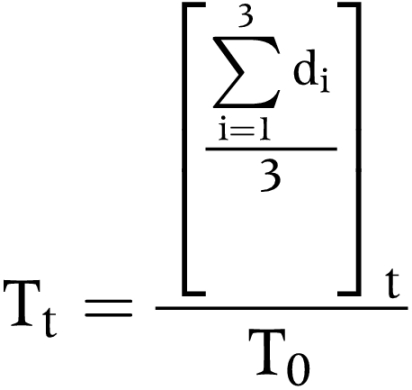 |
Statistics
Results are expressed as mean (SEM). Comparison of differences used the unpaired two tailed Student’s t test with a significance level of 0.05.
RESULTS
Immunofluorescence studies showed high levels of Mcl-1 (fig 1B, D ▶) and Bcl-XL (fig 1A, C ▶) expression in Egi-1 and Tfk-1.
Figure 1.

Flow cytometry of cell lines Egi-1 and Tfk-1 showing expression of Mcl-1 and Bcl-XL (open curve) against controls (shaded curve). Tfk-1 cells expressed high levels of both Mcl-1 (B) and Bcl-XL (A). Egi-1 cells had a similar profile (C, D).
Egi-1 and Tfk-1 cells were found to be markedly resistant in vitro to the apoptosis inducing effects of chemotherapy (fig 2C ▶) and radiotherapy (fig 2D ▶). Seventy two hours following exposure of Tfk-1 cells to 1 Gy of x ray irradiation there were only 28% of cells undergoing apoptosis (fig 2D ▶). This poor response was discovered to be by no means dependent on the form of DNA damage utilised. Both cells lines were resistant to apoptosis induction but Egi-1 appeared more robust than Tfk-1.
Figure 2.

Ability of Pk11195 to facilitate the response of cholangiocarcinoma cells to DNA damaging therapies. Cells were stained with the mitochondrial probe prior to analysis by flow cytometry. The number of cells undergoing mitochondrial permeability transition (MPT/apoptosis) is expressed as a fraction (0 to 1 where 1=100%) of the total number of cells analysed. (A, B) Apoptosis in Tfk-1 and Egi-1 cells, respectively, following a five minute (5′) exposure to ultraviolet (UV) irradiation alone or with addition of Pk11195. (C) Effect of 10 μM Vp16 induced MPT in Tfk-1 cells over 96 hours. In the presence of Pk11195 the response of Tfk-1 to Vp16 was enhanced over time. (D) Effect of 500 cGy and 1000 cGy x ray irradiation on Tfk-1 cells with or without the enhancing effects of Pk11195.
Addition of 75 μM Pk11195, the mBzR and Bcl-2 antagonist, significantly altered the kinetics of ΔΨm dissipation and apoptosis in both cells lines in response to chemotherapy (fig 2C ▶), UV, and x ray irradiation (fig 2A, B, D ▶). The 28% fraction of apoptosis of Tfk-1 at 72 hours following 1 Gy x ray irradiation mentioned above became 92% following addition of Pk11195 (fig 2D ▶). With addition of Pk11195 the percentage of Egi-1 cells undergoing apoptosis at 48 hours following exposure to UV irradiation rose from 26% to 53% (fig 2B ▶). The magnitude of the Pk11195 effect was dependent both on the dose of the apoptosis stimulator used and the time the experiment was analysed following treatment (fig 2 ▶). This effect is consistent with an apoptosis enhancing effect of Pk11195 on both Egi-1 and Tfk-1 cells. To verify that the mitochondrial events measured lead to caspase activation, we performed the caspase dependent experiment annexin V assay following treatment and found an identical dose and time dependent rise in phosphotidylserine expression (fig 3A ▶). Pk11195 75 μM on its own produced no cytotoxic effects on either cell line over a 96 hour period (fig 3B ▶). Cholangiocarcinoma cells took up the mBzR probe, as shown using NBD-FGIN-1–27 analogue30: the granular cytoplasmic pattern of distribution along with colocalisation with the mitochondrial fluorochrome CMXRos in both TFk-1 (fig 3C, D ▶) and Egi-1 (not shown) confirmed mitochondrial targeting.
Figure 3.


(A) Pk11195 increased the proportion of cells expressing phosphotidylserine after ultraviolet (UV) irradiation at 48 and 72 hours in Tfk-1, as shown using the fluorescein isothiocynate annexin V assay. (B) Pk11195 75 μM alone had no intrinsic cytotoxic effect on Tfk-1 over 96 hours. (C, D) Detection of mitochondrial benzodiazepine receptor using the fluorescent ligand 7-nitro-2, 1, 3-benzoxadiazol-4-yl derivative (NBD FGIN-1–27). Fluorescent images showing granular cytoplasmic staining of NBD FGIN-1–27 (green) in Tfk-1 (C). An identical staining pattern of the cell when incubated with the lipophilic fluorochrome chloromethyl-X-rosomine (CMXRos) (red) in Tfk-1 (D) confirmed mitochondrial binding.
In vivo, xenografts grown on mice treated with Vp16 alone on two separate occasions failed to respond to Vp16, with a growth pattern similar to control mice (fig 4A ▶). However, mice simultaneously treated with Pk11195 in addition to VP16 showed both growth slow down and regression of xenografts (fig 4A ▶). This effect was reproduced when mice were treated for a second time (fig 4A ▶). Similarly, Egi-1 xenografts exhibited a similar attenuation in growth over the 72 hours of treatment (fig 4B ▶).
Figure 4.

Growth curves of Tfk-1 xenografts in the three treatment groups over the 35 day experiment. The antitumour activity of Vp16 on the xenografts was facilitated in the presence of Pk11195. Animals were treated for a 72 hour period during each of the two sensitising schedules. The effects of Vp16 on xenograft growth were altered by addition of Pk11195 to the treatment regimen.
DISCUSSION
This study has demonstrated that the human cholangiocarcinoma cell lines Tfk-1 and Egi-1 express the antiapoptotic proteins Bcl-XL and Mcl-1. We have previously reported expression of the antiapoptotic proteins Bcl-XL and Mcl-1 in preserved human cholangiocarcinoma tissue.19 There have been other studies that have investigated resected human cholangiocarcinoma tissue and show expression of antiapoptotic proteins.20–22 These proteins are known for their ability to prevent cellular apoptosis both in vitro and in vivo in certain cell types.11–18 However, their effect on cholangiocarcinoma cell apoptosis has not been investigated.
Chemotherapy and radiotherapy are widely used in the treatment of many solid and haematological malignancies. In vitro and in vivo experiments into the behaviour of cancer cells following exposure to such forms of cancer therapy show induction of apoptosis as their vital mode of action. There are no studies on the response of human cholangiocarcinoma cells to chemotherapy and radiotherapy in vitro. This study has monitored the response of two cholangiocarcinoma cells lines to both chemotherapy and radiotherapy by measuring loss of inner mitochondrial potential as a rapid and efficient method of quantifying apoptosis at the single cell level.26–29 The low number of cells undergoing apoptosis confirms that in vitro cholangiocarcinoma cells are resistant to the apoptosis inducing effects of chemotherapy and radiotherapy.
The magnitude and rate of apoptosis following treatment of human cholangiocarcinoma cells with chemotherapy, UV, or x ray irradiation was increased in this study by simultaneous addition of the Bcl-2 and mBzR antagonist Pk11195. This molecule targets the mitochondria and functionally antagonises Bcl-2-like proteins through a mechanism not yet clearly understood.23 The ability of 75 μM Pk11195 to sensitise Bcl-XL and Mcl-1 expressing cholangiocarcinoma cells to apoptosis would suggest that antiapoptotic proteins expressed by cholangiocarcinoma cells are important in the known resistance to treatment, at least in vitro. That Pk11195 in combination with etoposide influenced xenograft growth demonstrates that it also possesses in vivo activity. Although there was some difference in the way the two cell lines responded to cytotoxic therapy, the ability of Pk11195 to increase the response to chemotherapy and radiotherapy in such a dose and time dependent manner supports a direct apoptosis enhancing effect by Pk11195.
Recent cancer therapies that target and antagonise the mitochondrial effects of antiapoptotic proteins or downregulation of antiapoptotic protein levels using antisense oligonucleotides have been shown to restore chemosensitivity in different haematological and solid tumours.31–33 Indirect strategies such as pharmacological agents that induce mitochondrial dysfunction through their effects on the PTPC are used in clinical practice. For example, 5 δ; aminolaevulinic acid is used as a prodrug to generate the endogenous ligand of the mBzR, protoporphyrin IX in photodynamic therapy.34–36 Arsenic trioxide has shown encouraging results in the treatment of patients with acute promyelocytic leukaemia37 and directly induces the mitochondrial release of cytochrome c through its interaction with the PTPC.38 The potential benefit of mitochondria targeted therapy in cholangiocarcinoma was highlighted in a report into the clinical responses observed following photodynamic therapy in unresectable cases of cholangiocarcinoma.39
It is possible and perhaps likely that there are other mechanisms besides antiapoptotic proteins involved in the resistance of cholangiocarcinoma cells to apoptosis. The clinical relevance of the finding from this study remains to be established. Mitochondrial targeting therapies using small molecules such as the mBzR antagonist Pk11195 in combination with chemotherapy or radiotherapy certainly provides a novel strategy for the treatment of cholangiocarcinoma.
Abbreviations
mBzR, mitochondrial benzodiazepine receptor
PTPC, permeability transition pore complex
FCS, fetal calf serum
FITC, fluorescein isothiocynate
CMXRos, chloromethyl-X-rosomine
UV, ultraviolet
DiOC6(3), dihexyloxalocardocyanine
SCID-NOD, severe combined immunodeficient non-obese diabetic
NBD FGIN-1-27 analogue, 7-nitro-2, 1, 3-benzoxadiazol-4-yl derivative
REFERENCES
- 1.Pazdur R, Royce ME, Rodriguez GI, et al. Phase II trial of docetaxel for cholangiocarcinoma. Am J Clin Oncol 1999;22:78–81. [DOI] [PubMed] [Google Scholar]
- 2.Sanz-Altamira PM, Ferrante K, Jenkins RL, et al. A phase II trial of 5-fluorouracil, leucovorin, and carboplatin in patients with unresectable biliary tree carcinoma. Cancer 1998;82:2321–5. [PubMed] [Google Scholar]
- 3.Ravry MJ, Omura GA, Bartolucci AA, et al. Phase II evaluation of cisplatin in advanced hepatocellular carcinoma and cholangiocarcinoma: a Southeastern Cancer Study Group Trial. Cancer Treat Rep 1986;70:311–12. [PubMed] [Google Scholar]
- 4.Poplin E, Roberts J, Tombs M, et al. Leucovorin, 5-fluorouracil, and gemcitabine: a phase I study. Invest New Drugs 1999;17:57–62. [DOI] [PubMed] [Google Scholar]
- 5.Junginger T, Ketterer K. Palliative therapy of gastrointestinal obstruction. Chirurg 1999;70:1397–401. [DOI] [PubMed] [Google Scholar]
- 6.Baines M, Oliver DJ, Carter RL. Medical management of intestinal obstruction in patients with advanced malignant disease. A clinical and pathological study. Lancet 1985;2:990–3. [DOI] [PubMed] [Google Scholar]
- 7.Eastman A. Apoptosis: a product of programmed and unprogrammed cell death. Toxicol Appl Pharmacol 1993;121:160–4. [DOI] [PubMed] [Google Scholar]
- 8.Dive C. Avoidance of apoptosis as a mechanism of drug resistance. J Intern Med Supplement 1997;740:139–45. [PubMed] [Google Scholar]
- 9.Hannun YA. Apoptosis and the dilemma of cancer chemotherapy. Blood 1997;89:1845–53. [PubMed] [Google Scholar]
- 10.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972;26:239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ibrado AM, Huang Y, Fang G, et al. Overexpression of Bcl-2 or Bcl-xL inhibits Ara-C-induced CPP32/Yama protease activity and apoptosis of human acute myelogenous leukemia HL- 60 cells. Cancer Res 1996;56:4743–8. [PubMed] [Google Scholar]
- 12.Ibrado AM, Liu L, Bhalla K. Bcl-xL overexpression inhibits progression of molecular events leading to paclitaxel-induced apoptosis of human acute myeloid leukemia HL-60 cells. Cancer Res 1997;57:1109–15. [PubMed] [Google Scholar]
- 13.Kim CN, Wang X, Huang Y, et al. Overexpression of Bcl-X(L) inhibits Ara-C-induced mitochondrial loss of cytochrome c and other perturbations that activate the molecular cascade of apoptosis. Cancer Res 1997;57:3115–20. [PubMed] [Google Scholar]
- 14.Yang E, Zha J, Jockel J, et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995;80:285–91. [DOI] [PubMed] [Google Scholar]
- 15.Kamesaki S, Kamesaki H, Jorgensen TJ, et al. bcl-2 protein inhibits etoposide-induced apoptosis through its effects on events subsequent to topoisomerase II-induced DNA strand breaks and their repair. Cancer Res 1993;53:4251–6. [PubMed] [Google Scholar]
- 16.Aranha GV, Folk FA, Greenlee HB. Surgical palliation of small bowel obstruction due to metastatic carcinoma. Am Surg 1981;47:99–102. [PubMed] [Google Scholar]
- 17.Clarke-Pearson DL, Chin NO, DeLong ER, et al. Surgical management of intestinal obstruction in ovarian cancer. I. Clinical features, postoperative complications, and survival. Gynecol Oncol 1987;26:11–18. [DOI] [PubMed] [Google Scholar]
- 18.Yang E, Zha J, Jockel J, et al. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995;80:285–91. [DOI] [PubMed] [Google Scholar]
- 19.Okaro AC, Deery AR, Hutchins RR, et al. The expression of antiapoptotic proteins Bcl-2, Bcl-X(L), and Mcl-1 in benign, dysplastic, and malignant biliary epithelium. J Clin Pathol 2001;54:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arora DS, Ramsdale J, Lodge JP, et al. p53 but not bcl-2 is expressed by most cholangiocarcinomas: a study of 28 cases. Histopathology 1999;34:497–501. [DOI] [PubMed] [Google Scholar]
- 21.Charlotte F, L’Hermine A, Martin N, et al. Immunohistochemical detection of bcl-2 protein in normal and pathological human liver. Am J Pathol 1994;144:460–5. [PMC free article] [PubMed] [Google Scholar]
- 22.Harnois DM, Que FG, Celli A, et al. Bcl-2 is overexpressed and alters the threshold for apoptosis in a cholangiocarcinoma cell line. Hepatology 1997;26:884–90. [DOI] [PubMed] [Google Scholar]
- 23.Hirsch T, Marzo I, Kroemer G. Role of the mitochondrial permeability transition pore in apoptosis. Biosci Rep 1997;17:67–76. [DOI] [PubMed] [Google Scholar]
- 24.Pastorino JG, Simbula G, Yamamoto K, et al. The cytotoxicity of tumor necrosis factor depends on induction of the mitochondrial permeability transition. J Biol Chem 1996;271:29792–8. [DOI] [PubMed] [Google Scholar]
- 25.Saijyo S, Kudo T, Suzuki M, et al. Establishment of a new extrahepatic bile duct carcinoma cell line, TFK-1. Tohoku J Exp Med 1995;177:61–71. [DOI] [PubMed] [Google Scholar]
- 26.Cai J, Yang J, Jones DP. Mitochondrial control of apoptosis: The role of cytochrome c. Biochim Biophys Acta Bioenerg 1998;2:139–49. [DOI] [PubMed] [Google Scholar]
- 27.Hirsch T, Marchetti P, Susin SA, et al. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 1573;15:1573–81. [DOI] [PubMed] [Google Scholar]
- 28.Hirsch T, Susin SA, Marzo I, et al. Mitochondrial permeability transition in apoptosis and necrosis. Cell Biol Toxicol 1998;14:141–5. [DOI] [PubMed] [Google Scholar]
- 29.Marchetti P, Castedo M, Susin SA, et al. Mitochondrial permeability transition is a central coordinating event of apoptosis. J Exp Med 1995;184:1155–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kozikowski AP, Kotoula M, Ma D, et al. Synthesis and biology of a 7-nitro-2,1,3-benzoxadiazol-4-yl derivative of 2-phenylindole-3-acetamide: a fluorescent probe for the peripheral-type benzodiazepine receptor. J Med Chem 1997;40:2435–9. [DOI] [PubMed] [Google Scholar]
- 31.Webb A, Cunningham D, Cotter F, et al. BCL-2 antisense therapy in patients with non-Hodgkin lymphoma. Lancet 1997;349:1137–41. [DOI] [PubMed] [Google Scholar]
- 32.Ackermann EJ, Taylor JK, Narayana R, et al. The role of antiapoptotic Bcl-2 family members in endothelial apoptosis elucidated with antisense oligonucleotides. J Biol Chem 1999;274:11245–52. [DOI] [PubMed] [Google Scholar]
- 33.Bloem A, Lockhorst H. Bcl-2 antisense therapy in multiple myeloma. Pathol Biol (Paris) 1999;47:216–20. [PubMed] [Google Scholar]
- 34.Svanberg K, Liu DL, Wang I, et al. Photodynamic therapy using intravenous delta-aminolaevulinic acid-induced protoporphyrin IX sensitisation in experimental hepatic tumours in rats. Br J Cancer 1996;74:1526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van den Boogert J, van Hillegersberg R, de Rooij FW, et al. 5-Aminolaevulinic acid-induced protoporphyrin IX accumulation in tissues: pharmacokinetics after oral or intravenous administration. J Photochem Photobiol B 1998;44:29–38. [DOI] [PubMed] [Google Scholar]
- 36.Verma A, Facchina SL, Hirsch DJ, et al. Photodynamic tumor therapy: Mitochondrial benzodiazepine receptors as a therapeutic target. Mol Med 1998;4:40–45. [PMC free article] [PubMed] [Google Scholar]
- 37.Soignet SL, Maslak P, Wang ZG, et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N Engl J Med 1998;339:1341–8. [DOI] [PubMed] [Google Scholar]
- 38.Larochette N, Decaudin D, Jacotot E, et al. Arsenite induces apoptosis via a direct effect on the mitochondrial permeability transition pore. Exp Cell Res 1999;249:413–21. [DOI] [PubMed] [Google Scholar]
- 39.Ortner MA, Liebetruth J, Schreiber S, et al. Photodynamic therapy of nonresectable cholangiocarcinoma. Gastroenterology 1998;114:536–42. [DOI] [PubMed] [Google Scholar]