

Abstract
The effects of pathogenic organisms on host intestinal epithelial cells are vast. Innumerable signalling pathways are triggered leading ultimately to drastic changes in physiological functions. Here, the ways in which enteric bacterial pathogens utilise and impact on the three major physiological functions of the intestinal epithelium are discussed: alterations in the structure and function of the tight junction barrier, induction of fluid and electrolyte secretion, and activation of the inflammatory cascade. This field of investigation, which was virtually non-existent a decade ago, has now exploded, thus rapidly expanding our understanding of bacterial pathogenesis. Through increased delineation of the ways in which microbes alter host physiology, we simultaneous gain insight into the normal regulatory mechanisms of the intestinal epithelium.
Keywords: tight junctions, enteric pathogens, ion transport, epithelium, inflammation
With the realisation that the intestinal epithelium is not merely a static barrier to the external environment, a wealth of information regarding eukaryotic and prokaryotic interactions has been gathered. It is now evident that this is a two way interaction involving a complex and dynamic “cross talk” between the species. A new language and understanding has emerged from these discoveries. Not only have we learned more about the mechanisms by which these pathogens usurp our defences and alter cellular functions, we better understand the normal physiological role of the intestinal epithelium. As the new dawn fades, it is imperative that we reflect upon what we have learned and contemplate what yet remains to be discovered. The purpose of this review is to give the reader an overview of the different mechanisms by which enteric pathogens induce epithelial responses. Three general categories of epithelial-pathogen interactions will be discussed: alterations in the structure and function of the tight junction barrier, induction of fluid and electrolyte secretion, and activation of the inflammatory cascade. It is noteworthy that while some pathogens primarily activate one of the above pathways, others such as Salmonella and Escherichia coli are well versed in all three languages.
TIGHT JUNCTION STRUCTURE AND BARRIER FUNCTION
Background
Enteric pathogens have devised several ways to disrupt the tight junctions (TJ) of epithelial cells. In general, this is achieved by either altering the cellular cytoskeleton or by affecting specific tight junction proteins. Tight junction regulation via the cytoskeleton may occur indirectly through changes in the perijunctional actomyosin ring or directly through changes in specific TJ proteins (fig 1 ▶). Disruption of specific TJ proteins can result from degradation by bacterial derived proteases or by biochemical alterations such as phosphorylation or dephosphorylation. These processes may lead to perturbation in epithelial functions such as establishment of electrochemical gradients as generated by active vectorial transport and paracellular permeability. Objectively, the function of TJs can be measured as a decrease in transepithelial electrical resistance (TER) and an increase in the paracellular flux of macromolecules such as mannitol. In the following section, examples of specific bacterial induced mechanisms of TJ disruption will be discussed.
Figure 1.
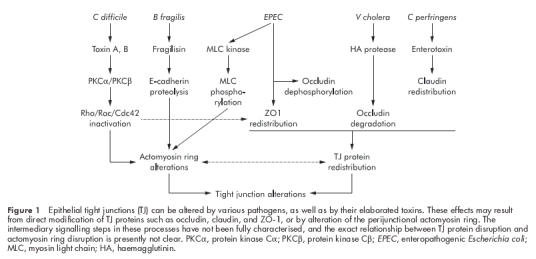
Epithelial tight junctions (TJ) can be altered by various pathogens, as well as by their elaborated toxins. These effects may result from direct modification of TJ proteins such as occludin, claudin, and ZO-1, or by alteration of the perijunctional actomyosin ring. The intermediary signalling steps in these processes have not been fully characterised, and the exact relationship between TJ protein disruption and actomyosin ring disruption is presently not clear. PKCα, protein kinase Cα; PKCβ, protein kinase Cβ; EPEC, enteropathogenic Escherichia coli; MLC, myosin light chain; HA, haemagglutinin.
Tight junction disruption due to alterations of the cytoskeleton
Clostridium difficile
Clostridium difficile is an anaerobic bacterium that causes antibiotic associated pseudomembranous colitis. The pathogenicity of this bacterium is attributable to elaboration of two enterotoxins, toxin A and toxin B. Exposure of eukaryotic cells to either of these toxins causes degradation of filamentous actin and increased levels of soluble actin, resulting in cell rounding.1,2 Functionally, decreased TER and increased flux of paracellular markers such as mannitol and raffinose occurs, indicating TJ disruption.2,3 These structural and functional changes appear to be due to toxin induced modifications of the Rho family of proteins.4,5 The Rho proteins (Rho, Rac, and Cdc42) regulate a wide variety of cell functions, such as cell shape, cell-cell interactions, and TJs, through organisation of the actin cytoskeleton.6,7 Toxins A and B monoglucosylate Rho, Rac, and Cdc42 using UDP-glucose as a co-substrate. This results in inactivation of the Rho GTPases and filamentous actin degradation.5,8 Loss of organisation in the perijunctional F-actin ring following inactivation of Rho by toxin A is believed to be a critical event in the mechanism of lowered TER and increased paracellular flux of solutes in toxin exposed intestinal epithelium.9 Associated with C difficile toxin induced depolymerisation of actin is the movement of TJ proteins such as ZO-1 and occludin away from the TJ and into the cytoplasm of the cell.9
“C difficile induced TJ disruption may be due to alterations in both the cytoskeleton and direct effects on TJ proteins”
Toxin A mediated decline in TER precedes changes in cell morphology and TJ ultrastructure,2 suggesting that C difficile may also affect TJ proteins directly. A recent study found that toxin A increased the redistribution of ZO-1 from TJs which corresponded with the activation of protein kinase Cα (PKCα) and protein kinase Cβ (PKCβ).10 Inhibition of PKCα and PKCβ with myristoylated PKCα/β blocked toxin mediated RhoA glucosylation, ZO-1 translocation, and cell rounding, indicting the proximal involvement of these signalling pathways. Together these findings suggest that C difficile induced TJ disruption may be due to alterations in both the cytoskeleton and direct effects on TJ proteins.
Escherichia coli
Enteropathogenic Escherichia coli (EPEC) has a complex biological arsenal. EPEC adheres to the surface of epithelial cells and induces accumulation of cytoskeletal proteins underneath the site of attachment to produce the characteristic attaching and effacing lesion.11 Expression of a type III secretion apparatus allows insertion of effector molecules into host cells. These proteins trigger a broad range of cellular events including alterations in electrolyte secretion, disruption of the TJ barrier, and inflammation. When T84 human intestinal epithelial monolayers are infected with EPEC, a dose and time dependent drop in TER occurs.12,13 Mutant strains deficient in the type III secretion have an attenuated effect on TER.13,14 Dual mannitol and 22Na+ flux studies confirmed that the permeability defect is at the level of the TJ.12 This occurs via a calcium and myosin light chain kinase (MLCK) dependent process.15 EPEC infection of intestinal epithelial cells stimulates phosphorylation of the 20 kDa myosin light chain (MLC20) by MLCK, accounting in part for the EPEC induced TJ disruption. MLC20 phosphorylation by MLCK stimulates cytoskeletal contraction, including that of the perijunctional actomyosin ring.16 This event causes a drop in TER and increase in paracellular permeability17,18 presumably by exerting tension on the cell membrane in the region of the zonula adherens where the perijunctional actomyosin ring inserts. This tension is transmitted to the TJ and thus increases paracellular permeability. Alternatively, cytoskeletal contraction may influence TJ proteins through direct interactions. In fact, EPEC has been shown to alter occludin, a transmembrane TJ protein that is important for barrier formation.19,20 Phosphorylation of occludin is required for its localisation to the TJ complex.21,22 Following infection by EPEC, occludin shifts from the TJ into the cytosol of the cell, an event that is accompanied by protein dephosphorylation.23 These findings correlate temporally with the decrease in TER. Reversal of these changes in occludin and the return of TER to baseline occurs following elimination of the infection with gentamicin. Furthermore, inhibition of serine/threonine phosphatases prevented EPEC induced changes in both occludin and TER, implicating their involvement in the regulation of TJ. Whether this phosphatase activity is host or bacteria derived remains to be discerned.
A genetically similar pathogen, enterohaemorrhagic E coli (EHEC) also lowers TER, increases the paracellular flux of mannitol, and alters ZO-1 distribution.24 The signalling pathways involved in this process include MLCK and conventional PKCs. In contrast, conventional PKCs do not appear to participate in the EPEC associated disruption of TJs.25 Therefore, although EHEC and EPEC cause similar functional perturbations, somewhat different mechanisms appear to be responsible.
Tight junction disruption due to bacterial derived proteases
Bacteroides fragilis
Bacteroides fragilis is an example of a bacterium that disrupts TJs by proteolytic degradation of TJ proteins. Diarrhoeic strains of B fragilis produce a 20 kDa extracellular toxin known as B fragilis enterotoxin (BFT) or fragilysin.26,27 This toxin is a metalloprotease with a zinc binding motif and has the ability to hydrolyse several proteins, including gelatin, fibrinogen, and actin in vitro.28 When applied to T84 monolayers, this toxin caused a decrease in TER and increase in paracellular permeability.29,30 This response was much more dramatic when toxin was applied to the basolateral, rather than the apical, surface of epithelial monolayers.29 Also, a transient increase in short circuit current (Isc), reflecting net ion transport, was seen when the toxin was applied basally but not apically.29 The alteration in TER and Isc did not coincide temporally suggesting that independent pathways lead to these effects. Morphologically, cells exhibited rounding and loss of microvilli.29,31 Staining for F-actin revealed dispersion from the normal location at the apical perijunctional ring and microvilli. Similar results were found with native human colonic tissue.32
“Bacteroides fragilis is an example of a bacterium that disrupts TJs by proteolytic degradation of TJ proteins”
Further study of BFT showed the site of proteolytic action to be the extracellular domain of the zonula adherens protein, E-cadherin.33 Immunofluorescent confocal microscopy of HT-29 cells showed complete loss of E-cadherin staining after one hour of BFT exposure. Western blot analysis using antibodies to the cytoplasmic domain of E-cadherin demonstrated the release of 28 and 33 kDa fragments from the intact 120 kDa protein on incubation with BFT. The predicted combined weight of the intracellular and transmembrane domains of E-cadherin is approximately 30 kDa, suggesting that cleavage occurs near the plasma membrane. In contrast with the initial ATP independent cleavage of E-cadherin, degradation to the 28 and 33 kDa fragments and the toxin induced morphological changes require cellular ATP. These findings suggest that the initial cleavage step is directly caused by BFT but that host proteases are responsible for the subsequent degradation of E-cadherin and alterations in actin. In theory, proteolysis of the intracellular domain of E-cadherin may disrupt the association of E-cadherin with β-catenin, which links E-cadherin to α-catenin, therefore causing actin disruption.
Vibrio cholera
Vibrio cholera is another enteric pathogen that alters barrier function through the elaboration of a protease. Like B fragilis, V cholera secretes a zinc binding metalloprotease, termed haemagglutinin protease (HA/P), which has the ability to act on a wide variety of substrates, including cholera toxin, El Tor cytolysin/haemolysin toxin, and CTXφ.34–36 A mutant strain of V cholera of the El Tor biotype (CVD110) that expresses HA/P, but not the toxins ctxA, zot, hylA or Ace, was shown to decrease TER and induce alterations in the distribution of F-actin and ZO-1 in cultured epithelial cells.37 Further investigation revealed that HA/P degrades occludin into two distinct fragments whereas ZO-1 remained intact.38 Immunofluorescent staining using antibodies against the intracellular domain of occludin showed that the cytoplasmic portion of this protein remained at the cell periphery. The specific bacterial metalloprotease inhibitor Zincov abolished occludin degradation and actin rearrangement. These findings suggest that the HA/P induced cleavage of occludin, alteration of ZO-1, and rearrangement of actin consequently result in TJ disruption.
Tight junction proteins as bacterial receptors
Clostridium perfringens
A more recently demonstrated and intriguing role served by TJ proteins in bacterial pathogenesis is that of a receptor for bacterial toxins. Clostridium perfringens is an anaerobic bacterium that is an important cause of food borne gastrointestinal illness and may also cause antibiotic associated diarrhoea.39–41 Disease is caused by C perfringens enterotoxin (CPE) producing strains.42 CPE has a C terminal binding domain, while the biologically active portion of the toxin is localised to the N terminus.43,44 Exposure of intestinal cells to CPE results in tissue damage followed by fluid and electrolyte secretion.45,46 After binding to the cell surface, CPE remains associated with the plasma membrane and increases membrane permeability.47 CPE membrane interactions lead to the formation of a series of complexes, 90, 135, 155, and 200 kDa in size, that are thought to be capable of forming pores in the apical plasma membrane.48,49 CPE binding correlates with the formation of the 90 kDa complex which serves as a precursor to the formation of a larger pore forming complex 49. Katahira et al demonstrated that a 20 kDa protein in Vero cells acted as the receptor for CPE, which they termed CPE-R.50 Two homologous proteins in human intestinal epithelial cells, initially called hCPE-R and hRVP-1, were subsequently determined to be members of the claudin family and therefore renamed claudin-3 and claudin-4.51,52 Further investigation determined that the 200 kDa CPE complex, but not the smaller complexes, also contain the TJ protein occludin.53 Certain point mutations in the N terminus of CPE abolished the formation of the larger complexes, whereas formation of the small 90 kDa complex was unaffected,53 suggesting that the presence of occludin in the larger complex is not simply due to occludin-claudin interactions. Structurally, CPE has been shown to induce disruption of TJ fibrils and remove claudin-4 from TJs when applied to the basolateral surface of rat liver cells.54 The C terminal portion alone is able to cause these same changes in MDCK cells, even though this fragment lacks cytotoxic activity.55 These morphological changes correlate temporally with decreased TER and increased paracellular flux.
Subsequent to the discovery that the TJ transmembrane proteins claudin-3 and -4 serve as receptors for the bacterial toxin CPE, it was reported that another TJ transmembrane protein, junctional adhesion molecule, functions as a receptor for reovirus.56 Also, the coxsackievirus and adenovirus receptor was recently recognised to be a transmembrane component of the TJ.57 In that this review is restricted to the impact of bacterial pathogens on intestinal function, the details of these studies will not be discussed here.
FLUID AND ELECTROLYTE SECRETION
Background
The intestinal epithelium has a remarkable capacity for fluid and electrolyte absorption. Approximately 8–9 litre of fluid enter the gut on a daily basis and all except for 100–200 ml/day are reabsorbed under normal conditions. Elaborate transport pathways, under the tight control of various neurotransmitters, hormones, inflammatory mediators, and intraluminal contents, exist to carry out this function. When this system becomes disrupted, as with bacterial infection for example, diarrhoea may result. The following section focuses on the biology of bacterial induced diarrhoea.
Epithelial electrolyte transport
Chloride secretion is the principle determinant of luminal hydration. With chloride secretion, paracellular movement of sodium follows. The resulting accumulation of luminal sodium chloride provides an osmotic gradient for the diffusion of water. Chloride secretion involves the concerted effort of several transporters. Chloride enters the basolateral membrane via the actions of the Na/K/2Cl cotransporter. Energy for this transport is largely provided by the inwardly directed sodium gradient set up by the basolaterally located Na, K-ATPase. K+ channels, also basally situated, provide the exit route for transported K+. The apically located cAMP dependent cystic fibrosis transmembrane conductance regulator (CFTR) is responsible for the majority of apical chloride secretion. There are also calcium activated chloride channels (CaCC) which secrete chloride in response to increased intracellular calcium levels.58 The mechanisms whereby pathogens stimulate chloride secretion via these two transporters are depicted in fig 2 ▶.
Figure 2.

Various enteric pathogens elicit a chloride secretory response by stimulating either of the two apical chloride secretory channels, cystic fibrosis transmembrane conductance regulator (CFTR) or calcium activated chloride channel (CaCC). In addition to the various mechanisms that directly activate these channels, proinflammatory processes induced by various pathogens also have the ability to stimulate chloride secretion. Thus induction of the proinflammatory cytokine interleukin 8 stimulates the transmigration of neutrophils to the lumen where they secrete 5′AMP. Conversion of this nucleotide to the secretagogue adenosine leads to chloride secretion. Similarly, induction of nitric oxide by various pathogens leads to cGMP dependent chloride secretion. DAG, diacylglycerol; TDH, thermostable direct haemolysin; TRH, TDH related haemolysin; NO, nitric oxide; PGE2, prostaglandin E2; Cox-2, cyclooxygenase 2; ETEC, enterotoxigenic E coli; EAEC, enteroaggregative E coli; PLC, phospholipase C; CaMKII, calmodulin dependent protein kinase; PIP2, phosphatidyl inositol 4,5-bisphosphate; IP3, inositol 1,4,5-trisphosphate.
Cyclic AMP mediated fluid secretion
Various pathogens, including V cholera, E coli, Salmonella, Campylobacter jejuni, Shigella dysenteriae, and Pseudomonas aeruginosa, mediate chloride secretion in a cAMP dependent fashion.59,60 This is usually accomplished through the elaboration of enterotoxins. The prototypical and best characterised of these toxins is the AB5 enterotoxin family. Cholera toxin from V cholera and the type I (LTI) and type II (LTII) heat labile enterotoxins from E coli are among this group. These toxins consist of an A subunit non-covalently bound to five identical B polypeptides. The B subunit forms a pentameric structure and is responsible for toxin binding to the cell surface. The receptor for the B subunit is the oligosaccharide domain of ganglioside receptors. Binding specificity is determined by the B subunit. For example, cholera toxin and LTI bind to ganglioside GM1, whereas the LTII variants LTIIa and LTIIb bind to GD1b and GD1a, respectively.61,62 The A subunit is the enzymatically active portion of the toxin. It contains a protease sensitive “nick site” and a disulphide bond. Cleavage of the nick site and reduction of the disulphide bond results in the release of two separate protein subunits, A1 and A2. The enzymatically active A1 subunit ADP-ribosylates the α-subunit of Gs, a member of the G protein family. ADP-ribosylation of Gsα causes it to dissociate from the membrane bound β γ Gs complex. Once free, it binds to and activates the catalytic subunit of adenylyl cyclase located on the basolateral membrane.63 The resulting increase in intracellular cAMP levels leads to the activation of cAMP dependent protein kinase and subsequently CFTR. The final result is stimulation of electrogenic Cl− secretion and massive diarrhoea. In fact, CFTR−/− mice do not secrete fluid on exposure to cholera toxin.64 Cholera toxin may also indirectly affect intestinal secretion by inducing the enteric nervous system to release increased levels of 5-hydroxytryptamine.65
Cyclic GMP mediated fluid secretion
Under physiological conditions, increased intracellular cGMP can also lead to phosphorylation and activation of CFTR by cGMP dependent protein kinase II, resulting in chloride and bicarbonate secretion.66 Guanylate cyclase is responsible for the generation of GMP. While several isoforms of transmembrane guanylate cyclase exist, this review will focus on guanylate cyclase C (GC-C), which is the receptor exploited by bacterial pathogens.
Guanylin and uroguanylin are endogenous small peptides that stimulate chloride and bicarbonate secretion in the intestine through activation of GC-C.67,68 Several bacteria elaborate heat stable toxins (ST) that share considerable homology with guanylin and uroguanylin.67–69 ST binding to GC-C increases intracellular cGMP and initiates a signalling cascade, leading to phosphorylation of CFTR followed by chloride and bicarbonate secretion.
STa from enterotoxigenic E coli shares 50% homology with guanylin and was the first toxin found to activate cGMP. Subsequently, numerous other pathogens have been found to elaborate ST-like toxins. Enteroaggregative E coli produce an ST-like toxin, called EAST1, which is functionally and structurally similar to guanylin.70,71 The EAST1 phenotype has also been identified with significant frequency in EHEC (88.0%), EHEC serogroup O157:H7 (100%), EAggEC (41–86.6%), EPEC (22–58.3%), enterotoxigenic E coli (41%), and Salmonella species (11.9%).72,73. The EAST1 gene of EHEC is chromosomally located whereas it is plasmid encoded in other E coli species. Yersinia enterocolitica, V cholerae (O1 and non-O1 strains), and Klebsiella pneumoniae also produce heat stable toxins that are involved in the diarrhoea induced by these organisms.74–76
Calcium mediated fluid secretion
Intracellular calcium levels are generally very low (approximately 100 nM) and are under tight physiological control. Changes in intracellular calcium concentrations are transient, even in the continued presence of agonist. Calcium is sequestered into three compartments, the extracellular space, mitochondria, and non-mitochondrial intracellular stores. A number of neurohormonal substances, including acetylcholine, serotonin, carbachol, and bradykinin, can increase intracellular calcium by altering the permeability of these stores.77–80 Binding of ligand to receptor results in activation of the membrane associated phospholipase C. This enzyme hydrolyses phosphatidyl inositol 4,5-bisphosphate (PIP2), releasing inositol 1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). IP3 then acts to increase intracellular calcium levels while DAG activates PKC.
“Vibrio parahaemolyticus is an important cause of gastroenteritis worldwide with the thermostable direct haemolysin (TDH) toxin being a major virulence factor”
The first human CaCC was cloned in 1998.81 These apically located channels are activated by increases in cytosolic calcium. Regulation of these channels is complex involving phosphorylation by the calmodulin dependent protein kinase CaMKII and possibly PKC.81,82 Vibrio parahaemolyticus is an important cause of gastroenteritis worldwide with the thermostable direct haemolysin (TDH) toxin being a major virulence factor. TDH positive strains cause haemolysis on Wagatsuma agar medium, known as the Kanagawa phenomenon. TDH has been shown to cause intestinal fluid secretion, cytotoxicity, and a dose dependent increase in intracellular calcium.83 In an experimental model, TDH caused increased Isc when applied to the luminal surface of colonic epithelial cells which was inhibited by 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid, an inhibitor of CaCC. Similarly, PKC inhibitors attenuated the increase in intracellular calcium and Isc caused by TDH, indicating that PKC induced phosphorylation is also involved.84 Kanagawa phenomenon negative Vibrio parahaemolyticus produce a TDH related haemolysin that also increases Cl− secretion and raises intracellular calcium similar to levels seen with TDH.85
Briefly, non-bacterial microbial pathogens, most notably rotavirus, a common cause of severe diarrhoea in children,86 also exploit a calcium based mechanism to stimulate intestinal secretion. This involves expression of the non-structural glycoprotein NSP4 which acts as a viral enterotoxin to elicit calcium dependent chloride secretion.87,88 NSP4 was shown to increase intracellular calcium through both release from intracellular stores and through the plasmalemma, mediated through activation of phospholipase C and IP3 mobilisation.89
Bacterial driven host derived factors that increase chloride secretion
In addition to bacterial toxins that function directly as secretagogues, bacterial infection of host intestinal epithelial cells can also upregulate the expression of host derived products that, through autocrine or paracrine effects, also stimulate intestinal, primarily chloride, secretion. Such secondary events are discussed in this section.
Galanin
Most recently, galanin has been recognised as a mediator of intestinal ion secretion in response to infection by enteric bacterial pathogens. Galanin is a neuropeptide released from the nerve endings of the enteric nervous system.90 In addition to its ability to modulate intestinal motility, galanin induces chloride secretion in human intestinal epithelium through activation of the galanin-1 receptor (Gal-1R).91,92 Interestingly, cloning of Gal-1R revealed the presence of putative nuclear factor κB (NFκB) binding motifs.93 Subsequent studies confirmed that Gal-1R expression is upregulated in an NFκB dependent fashion and that such upregulation plays a role in fluid secretion on bacterial infection, as well as in DSS induced colitis.94,95
“The enteric pathogens EHEC, EPEC, ETEC, Salmonella typhimurium, and Shigella flexneri were all shown to increase Gal-1R expression in vitro and in vivo”
Specifically, the enteric pathogens EHEC, EPEC, ETEC, Salmonella typhimurium, and Shigella flexneri were all shown to increase Gal-1R expression in vitro and in vivo. The chloride secretory response to galanin was consequently accentuated following infection by these pathogens in both model systems and was significantly reduced by Gal-1R antibody as well as in Gal-1R−/− mice.96 Further studies demonstrated the galanin induced secretory response to be a calcium mediated event although the details of these stimulatory pathways are yet to be defined.94,96 Further investigation into this process has revealed an even broader role for Gal-1R expression and activation in secretory diarrhoea.97,98
Nitric oxide
Another host derived bacterial induced secretory agent is nitric oxide (NO). NO has several important functions in the intestinal epithelium, including regulation of barrier function and antimicrobial activity.99–101 NO is synthesised by the conversion of l-arginine to l-citrulline by nitric oxide synthase (NOS).102 Recently it has been shown that NO is capable of stimulating intestinal epithelial chloride secretion by increasing intracellular cGMP levels.103 Several enteric pathogens, and even lipopolysaccharide (LPS) alone, have been shown to activate inducible NOS (iNOS).104 The invasive bacteria S dublin, S flexneri, enteroinvasive E coli (EIEC), and Shigella flexneri, but not non-pathogenic non-invasive organisms, are capable of inducing iNOS expression.98,105–107 Infection by these pathogens was shown to increase intracellular cGMP and electrogenic chloride secretion.98 These responses were attenuated by inhibitors of NFκB and tyrosine kinases, thereby implicating their involvement in this pathway.107
Prostaglandins
Prostaglandins are the products of arachidonic acid metabolism through the cyclooxygenase pathway. Prostaglandin E2 (PGE2) production is catalysed by the enzyme prostaglandin H synthase (PGHS) and stimulates chloride secretion in epithelial cells.108,109. The invasive bacteria Shigella dysenteriae, Salmonella dublin, Salmonella typhi, Salmonella typhimurium, Yersinia enterocolitica, and EIEC, but not non-pathogenic bacteria, induced the expression of PHGS-2 and its products PGE2 and PGF2α, resulting in chloride secretion.110 Similar results have been found with other pathogens including Cryptosporidium parvum, Helicobacter pylori, and Entamoeba histolytica.111–113 Cyclooxygenase 2 (COX-2) induction by S dublin and EIEC also resulted in increased prostaglandin production which, in turn, contributed to chloride secretion.98 It is clear therefore that the host cell response to infection by enteric pathogens is capable of contributing to the intestinal secretory process, and ultimately diarrhoea, through a variety of mechanisms. The advantage to the host is the potential “flushing” of the intestinal lumen and resultant clearing of pathogenic microbes. The advantage of this process to the microbe is the increased potential for transmission to additional hosts.
EPITHELIAL RESPONSE TO ENTERIC PATHOGENS: ACTIVATION OF THE INFLAMMATORY CASCADE
Background
In addition to the effects on TJs and water and electrolyte secretion, intestinal pathogens are also capable of inciting inflammation in the gastrointestinal mucosa. While there are several different mechanisms by which this may occur, the common final response is the release of cytokines, chemokines, and the recruitment of inflammatory cells. The means by which this process is carried out is complex, and therefore not completely understood. The recently described roles of the NOD proteins, toll-like receptors (TLRs), CD14, and pathogen associated recognition patterns have certainly shed light on these complicated interactions and responses. This particular topic has recently been reviewed114 and so will not be covered here. Instead, the focus of this review will be limited to that of signalling pathways that activate the key regulator of inflammation, NFκB.
“In addition to the effects on TJs and water and electrolyte secretion, intestinal pathogens are also capable of inciting inflammation in the gastrointestinal mucosa”
There is tremendous divergence in the signalling pathway systems that are activated not only by different pathogens but by a single organism. For example, EPEC infection activates NFκB, mitogen activated protein (MAP) kinases, tyrosine kinases, and PKCs, all of which are involved in inducing the inflammatory response. Detailed knowledge of the signalling molecules involved in this process and their inter-relationship will not only allow for a better understanding of the interaction between bacteria and host, but will also provide insight into clinical situations in which inflammation goes unchecked, as in inflammatory bowel disease. This section begins with an overview of the mediators in gut inflammation and the messenger systems involved. Finally, the mechanisms by which inflammation is induced by specific bacterial pathogens will be discussed.
Inflammatory mediators
Enteric pathogens elicit expression of a characteristic profile of cytokines from epithelial cells that recruit effector cells to the site to clear the infecting organisms. The mucosal immune system is quite remarkable in its ability to protect against invasion by pathogens, yet not respond to the commensal bacterial flora or dietary antigens. The means by which enteric pathogens initiate inflammatory signals is complex and just beginning to be elucidated. After bacterial adherence or invasion, epithelial cells respond by secreting or expressing a characteristic pattern of cytokines, adhesion molecules, and major histocompatibility complex (MHC) class II molecules. These molecules, whose expression is regulated by various nuclear transcription factors and protein kinases, recruit a wide variety of effector cells, including neutrophils, monocytes, lymphocytes, and eosinophils to the site of infection.
Epithelial control of neutrophil transepithelial migration is mediated by the polarised secretion of distinct chemokines. For example, interleukin (IL)-8 is secreted basolaterally by the intestinal epithelium in response to pathogenic bacteria or specific proinflammatory cytokines (fig 3 ▶). This polarised secretion leads to the creation of IL-8 gradients that are largely responsible for neutrophil migration through the extracellular matrices of model epithelia and to the apical surface of epithelial cells.115 Once at the subepithelial surface, further signals are needed to complete transepithelial migration such that neutrophils will be strategically positioned adjacent to the pathogens. One such signal recently described is an apically secreted factor in response to S typhimurium infection, termed pathogen elicited epithelial derived chemoattractant (PEEC). This appears to be purely chemotactic to neutrophils and does not induce superoxide production or degranulation.116 Further characterisation of this molecule is awaited.
Figure 3.
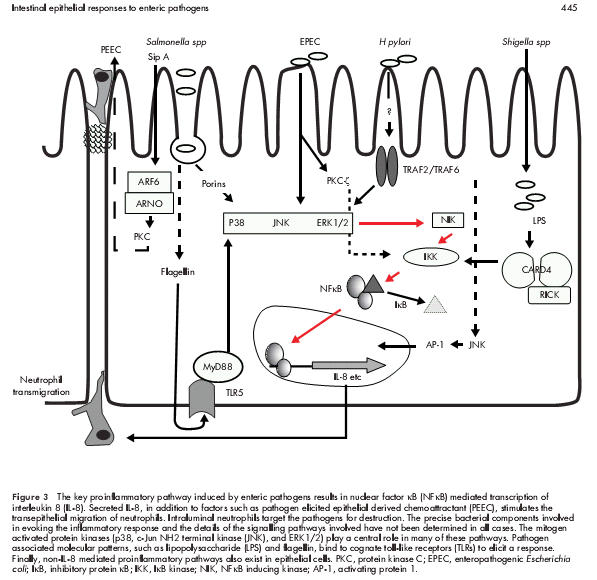
The key proinflammatory pathway induced by enteric pathogens results in nuclear factor κB (NFκB) mediated transcription of interleukin 8 (IL-8). Secreted IL-8, in addition to factors such as pathogen elicited epithelial derived chemoattractant (PEEC), stimulates the transepithelial migration of neutrophils. Intraluminal neutrophils target the pathogens for destruction. The precise bacterial components involved in evoking the inflammatory response and the details of the signalling pathways involved have not been determined in all cases. The mitogen activated protein kinases (p38, c-Jun NH2 terminal kinase (JNK), and ERK1/2) play a central role in many of these pathways. Pathogen associated molecular patterns, such as lipopolysaccharide (LPS) and flagellin, bind to cognate toll-like receptors (TLRs) to elicit a response. Finally, non-IL-8 mediated proinflammatory pathways also exist in epithelial cells. PKC, protein kinase C; EPEC, enteropathogenic Escherichia coli; IκB, inhibitory protein κB; IKK, IκB kinase; NIK, NFκB inducing kinase; AP-1, activating protein 1.
Interestingly, once the transepithelial migration of neutrophils has occurred and these cells are luminally situated, they also contribute to intestinal epithelial chloride secretion. Neutrophils release 5′AMP, which is converted to adenosine by the apical membrane enzyme 5′ ectonucleotidase (CD73).117 Interaction of adenosine with the intestinal adenosine receptor A2b stimulates the secretion of Cl− through a Gαs and cAMP dependent mechanism.118
Signal transduction pathways
Nuclear factor κB
NFκB clearly plays a pivotal role in intestinal inflammation. There is evidence that it also regulates apoptosis in certain cell lines.119,120 NFκB controls the expression of essentially all proinflammatory cytokines, chemokines, immune receptors, and cell surface adhesion molecules including IL-1β, tumour necrosis factor α (TNF-α), IL-6, IL-8, IL-12, iNOS, intercellular adhesion molecule 1, vascular cell adhesion molecule 1, T cell receptor α, and MHC class II molecules. Both invasive and non-invasive enteric pathogens, but not commensal flora, trigger the inflammatory cascade through activation of NFκB. The clinical importance of this is illustrated by the presence of activated NFκB in the crypt cells of the intestine of patients with Crohn’s disease and ulcerative colitis. The magnitude of this activation correlates closely with disease activity as does the expression of cytokines including IL-8 and IL-6.121–123
“NFκB clearly plays a pivotal role in intestinal inflammation”
Inactive NFκB exists as either a hetero- or homodimer in the cytoplasm, bound to one of the members of the inhibitory IκB family of proteins. IκB kinase (IKK) phosphorylates serine residues on IκB, targeting it for ubiquitination by proteasomes, thus liberating NFκB. Dissociation of NFκB from IκB unmasks the nuclear translocation domain of NFκB, allowing it to move into the nucleus and activate transcription of a variety of genes, including those involved in inflammation.
Recent studies suggest that phosphorylation of the NFκB RelA/p65 subunit is also necessary for induction of NFκB dependent transcription. The kinases responsible for this phosphorylation event have only recently been identified. For example, TNF-α induced p65 phosphorylation is mediated by casein kinase II.124,125 Phosphorylation of p65 by protein kinase A is required for efficient binding to the transcriptional activator protein CBP.126 PKCζ127 as well as phosphotidylinositol 3 kinase128 has also been demonstrated to phosphorylate p65.
The extracellular signals responsible for activation of NFκB are many and are still being defined but activation of IKK is the common end point of many of these stimuli. IKK is activated by phosphorylation but the molecules that perform this task in vivo are unknown. Certain MAP kinases (MEKK1, MEKK2, MEKK3, and NIK) have been shown to phosphorylate IKK in vitro but their role in in vivo regulation has yet to be defined.
Activation of NFκB by cytokines, such as TNF-α and IL-1, is initiated by their interaction with cognate receptors located in the cell membrane. In these situations, NFκB inducing kinase (NIK) communicates with the intracellular domain of these receptors triggering a complex array of downstream signalling events that ultimately activate IKK and, in turn, NFκB. However, there is increasing evidence to suggest that bacteria employ cytokine independent pathways to activate NFκB.129 The mechanisms by which bacteria stimulate the NFκB pathway, an area of intense interest, are discussed below.
MAP kinases
The MAP kinase (MAPK) family is involved in many signal transduction pathways, including the inflammatory response. While this family may be involved in membrane, cytosolic, cytoskeletal, and nuclear processes, they commonly target transcription factors that impact a diverse array of cellular functions, including cytokine production,130,131 There are three parallel MAP kinase cascades, ERK, JNK, and p38 that can be simultaneously or independently activated. The cascade is initiated by activation of MAPK kinase kinase (MAPKKK). One of the best defined means of MAPKKK activation is through receptor tyrosine kinases.132 Receptor autophosphorylation caused by ligand binding induces the recruitment of SH2 containing adaptor proteins, which subsequently recruit guanine nucleotide exchange factors near the membrane, promoting GTP binding to Ras. GTP bound Ras binds the protein kinases Raf-1 and B-Raf thus increasing their protein kinase activity. Raf-1 and B-Raf then phosphorylate MAPKK, also called MAP/ERK kinases (MEK), which subsequently phosphorylates and activates the MAP kinases.
The pathways by which ERKs are activated have been studied in a number of cell systems. Signals from many receptors lead to activation of ERK through Ras-Raf1-MEK.130 A Ras independent pathway that involves Raf1 and PKCδ has also been described.133 In addition, the ERK pathway can be activated by PKCζ through its interaction/activation of MEK.134–138 Downstream, ERK may be involved in the regulation of NFκB through MEK, which has been shown to activate both IKK-α and IKK-β.139,140
Protein kinase C
The PKC family consists of 11 identified members divided into three groups based, in part, on their biochemical and sequence homologies.141–143 The conventional PKCs (cPKC-α, β1, β2, and γ) are activated in a calcium and DAG dependent manner. Novel PKCs (nPKC-δ, ɛ, η, θ, and μ) are calcium independent but DAG dependent. Atypical PKCs (aPKC-ζ and λ/ι) are DAG and calcium independent and, in contrast with conventional and novel PKCs, do not respond to phorbol esters. The different isotypes have signal and tissue specificity. For example, PKCζ is involved in signal transduction pathways critical for cell proliferation and survival.134,144,145 This is most likely by activating ERK and NFκB transcription factors.136,137,146–152 Studies have demonstrated that the atypical PKCs are required for TNF-α induced activation of NFκB.153 Additionally, PKCζ has been shown to activate IKKβ in TNF-α stimulated cells by enhancing its interaction with IKK.135,149,154–156 Alternatively, PKCζ can phosphorylate the p65 subunit of NFκB thereby increasing its DNA binding activity.127
In summary, the above overview highlights the complex and redundant nature of the regulation of NFκB activation. The involvement of this singular transcription factor in so many critical cellular responses mandates such tight yet overlapping levels of regulation.
Bacterial activation of inflammation and secondary messengers
Numerous enteric pathogens activate the inflammatory response. Whether this is advantageous to the bacterium or the host remains controversial. In most cases it is probably a combination of both. Bacteria may use the host cellular apparatus for invasion, attachment, or synthesis of bacterial proteins. In turn, this exploitation of mammalian molecules usually results in the activation of mechanisms to clear the invader. Pathogens utilise many inducers of the inflammatory cascade. Such redundancy guarantees that inflammation is the outcome. The above mentioned signalling pathways and kinases are commonly utilised to achieve this goal.
“Invasion itself is not necessary for induction of inflammation”
Infection of epithelial monolayers with strains of invasive bacteria such as Salmonella dublin, Shigella dysenteriae, Yersinia enterocolitica, Listeria monocytogenes, and enteroinvasive E coli results in the expression of proinflammatory cytokines such as IL-8, monocyte chemotactic protein 1, granulocyte macrophage-colony stimulating factor, and TNF-α.157,158 In contrast, cytokine gene expression is not induced after infection of colon epithelial cells with the non-invasive bacteria Streptococcus bovis, Enterococcus faecium, E coli serotype 029, or the non-invasive protozoan parasite Giardia lamblia.157 However, invasion itself is not necessary for induction of inflammation. Inhibition of invasion of S dublin and S typhimurium does not abolish degradation of IκB, activation of NFκB, or IL-8 secretion but bacterial adhesion is required.159,160
Specific pathogens
Salmonella typhimurium
Salmonella typhimurium is a prototypical bacterium which induces a robust inflammatory response during infection. This is mostly mediated through basolateral secretion of IL-8 and recruitment and activation of neutrophils.161 IL-6 however is apically secreted and may also be involved in the inflammatory response.162 Recently, it has been described that soluble flagellin is translocated across epithelial cells to the basolateral membrane where it activates NFκB, elicits IL-8 secretion, and induces iNOS expression.160,163,164 The polarity of flagellin activity is due to the exclusively basolateral localisation of its receptor TLR5.165 Flagellin engagement of TLR5 also results in the recruitment of the adaptor protein MyD88 to the membrane. These effects are independent of the invasive phenotype of Salmonella.166
“Salmonella typhimurium induced activation of NFκB and IL-8 secretion is through calcium dependent phosphorylation of IκBα”
Salmonella typhimurium induced activation of NFκB and IL-8 secretion is through calcium dependent phosphorylation of IκBα. Inhibition of increased intracellular calcium abolishes IL-8 secretion in response to infection.167 The MAP kinases ERK, JNK, and p38 are also activated in epithelial cells infected with Salmonella typhimurium and may be involved in the full expression of the inflammatory response. This activation is dependent on invasion associated type III secretion. Inhibition of p38 by SB203580 reduces S typhimurium induced IL-8 secretion.168 Other investigators have found that although ERK1/2 and JNK are activated in Caco-2 cells by S typhimurium infection, inhibition of their activation by bromelain does not prevent IL-8 secretion, invasion of Caco-2 cells, or the drop in transepithelial resistance caused by Salmonella. In fact, bromelain paradoxically causes a synergistic increase in IL-8 production with Salmonella infection.169 In a monocyte cell line (U937), Salmonella typhimurium porins activate the transcription factors activating protein 1 (AP-1) and NFκB through activation of the MAP kinase cascade. There is a porin mediated increase in Raf-1 phosphorylation, which is accompanied by phosphorylation of MAPK kinase ½12 (MEK1/2), p38, ERK1/2, and JNK. Inhibition of p38, but not MEK1 or Raf-1, attenuates porin mediated activation of AP-1 and NFκB.170 Collectively, this information provides evidence that although the MAP kinases are activated in S typhimurium infection, perhaps only p38 is involved in modulation of IL-8 expression. The role of the remaining activated MAP kinases remains to be determined. In vitro studies suggest that Salmonella invasion can be uncoupled from its ability to induce PMN transmigration. Consistent with this, the release of PEEC does not require bacterial entry but does require SPI-1. The SPI-1 encoded protein SipA was shown to be required and sufficient for inducing PMN transmigration but not for bacterial entry.171 Following infection, SipA induces the recruitment of the GTPase ARF6 and its guanine exchange factor ARNO to the apical membrane.172 Activation of ARF6 results in activation of phospholipase D at this site. The phosphatidic acid generated by this enzyme is converted to DAG which then recruits PKC to the apical membrane. This in turn triggers a signalling cascade that results in the release of PEEC into the lumen. PEEC, a small stable molecule (<1 kDa), induces a signal transduction cascade involving the G protein Gi, and elevation of intracellular calcium.173 Thus ARF6 appears to act independently of the IL-8 pathway.
Enteropathogenic Escherichia coli
The pathogenesis of EPEC involves complex host-bacterial interactions including intimate attachment, type III secretion, and delivery of effector molecules into host cells.174 Like Salmonella, EPEC induces IL-8 secretion through activation of NFκB. However, unlike Salmonella, this occurs in a calcium independent manner.14,175,176 EPEC also activates the MAP kinases ERK1/2, p38, and JNK. Inhibition of these kinases does not however affect EPEC induced disruption of TJs or actin accumulation beneath the site of bacterial attachment, but does significantly decrease IL-8 secretion.176,177 PKCζ also appears to be involved in EPEC induced inflammation by interacting with and activating IKK.129 EPEC has also been shown to stimulate other signal transduction pathways in IEC including tyrosine kinase,178 phosphatidylinositol 3-kinase,179 and conventional PKCs.180 The contribution of these signalling pathways to EPEC pathogenesis or the host response has not been defined.
Clostridium difficile
Clostridium difficile is the pathogenic organism responsible for pseudomenbranous colitis and some milder forms of antibiotic associated diarrhoea. C difficile causes dramatic inflammation through the elaboration of several toxins. Toxin A for example has been shown to induce IL-8 secretion in intestinal epithelial cells.181,182 Although toxins A and B have both been shown to induce IL-8 secretion in monocytes through activation of NFκB, the pathways involved in epithelial cells remain to be characterised.183
Shigella
Shigella species infect the basolateral membrane of epithelial cells and induce the elaboration of a broad range of cytokines, including: IL-1, TNF-α, IL-6, IL-8, IL-4, IL-10, interferon γ, TNF-β, and transforming growth factor β.184,185 Epithelial cells are responsible for the secretion of IL-6 and IL-8, with the subsequent recruitment of neutrophils.185,186 Shigella induced IL-8 secretion results from NFκB activation.187 This involves activation of NIK, IKK1/2, and possibly ERK1/2 in an LPS dependent manner.187,188 Shigella are invasive pathogens that localise to host cell cytosol. It was recently shown that epithelial cells that are refractory to extracellular bacterial LPS respond to this molecule when it is delivered directly into the cytosol.189 LPS delivered into the cytosol by invasive Shigella flexneri was able to stimulate NFκB, possibly via the JNK pathway (see above). This response was dependent on a cytosolic protein called CARD4/Nod1 and was blocked by dominant negative CARD4. On S flexneri infection, CARD4 oligomerises and forms a transient complex with RICK and the IKK complex. It is possible that other CARD-4-like proteins may play a role in the recognition of, and response to, other bacterial determinants.
Helicobacter pylori
Helicobacter pylori is a gram negative bacteria capable of colonising the gastric mucosa. Colonisation induces chronic gastritis and increases the risk of peptic ulcer disease, gastric adenocarcinoma, and MALT lymphomas.190,191 The risk of such complications may be related to the presence of the cag (cytotoxin associated antigen) pathogenicity island in certain strains of H pylori.192–194 CagA positive H pylori have a much more profound effect on IL-8 secretion and mucosal inflammation than cagA negative strains.194–196 H pylori induced IL-8 secretion occurs through activation of NFκB and AP-1 and requires proteins encoded by the cag pathogenicity island.197–199 The upstream activators of NFκB appear to involve TRAF2 and TRAF6 activation of NIK with the subsequent activation of IKK, leading to IκB degradation and NFκB release.200 The MAP kinase pathway also plays a key role. CagA positive H pylori rapidly activates ERK, p38, and JNK MAP kinases.201 Simultaneous inhibition of MEK-1 and p38 completely abolishes IL-8 production but has no effect on NFκB activation suggesting that alternative mechanisms are responsible. The MAP kinase JNK is involved in the activation of AP-1 which along with NFκB may be required for full IL-8 gene expression.198 In addition to IL-8 secretion, H pylori also induces epithelial cell derived neutrophil activating protein 78 (ENA-78).202 However, unlike IL-8, ENA-78 expression does not appear to be correlated with the presence or absence of the cagA phenotype.
SUMMARY
The effects of pathogenic organisms on host intestinal epithelial cells are vast. Innumerable signalling pathways are triggered leading ultimately to drastic changes in physiological functions. We have attempted in this review to touch on ways in which enteric bacterial pathogens utilise and impact upon the three major physiological functions of the intestinal epithelium: the TJ barrier, active ion transport, and induction of the inflammatory response. This field of investigation, which was virtually non-existent a decade ago, has now exploded, thus rapidly expanding our understanding of bacterial pathogenesis. Through increased delineation of the ways in which microbes alter host physiology, we simultaneous gain insight into the normal regulatory mechanisms of the intestinal epithelium.
Acknowledgments
Research in the Hecht laboratory is supported by grants from the National Institutes of Health #DK50694 and DK58964 (GH) and Merit Review and Research Enhancement Awards from the Department of Veterans Affairs (GH). SDS is supported by a Research Fellowship Award from the Crohn’s and Colitis Foundation of American. We would like to thank Ms Alejandra Mendoza for her expert assistance in the preparation of this manuscript.
Abbreviations
TJ, tight junctions
TER, transepithelial electrical resistance
PKC, protein kinase C
PKCα, protein kinase Cα
PKCβ, protein kinase Cβ
EPEC, entero-pathogenic Escherichia coli
MLCK, myosin light chain kinase
EHEC, enterohaemorrhagic Escherichia coli
BFT, B fragilis enterotoxin
Isc, short circuit current
HA/P, haemagglutinin protease
CPE, C perfringens enterotoxin
CPE-R, C perfringens enterotoxin receptor
CFTR, cystic fibrosis transmembrane conductance regulator
CaCC, calcium activated chloride channel
GC-C, guanylate cyclase C
ST, stable toxins
DAG, diacylglycerol
IP3, inositol 1,4,5-trisphosphate
TDH, thermostable direct haemolysin
Gal-1R, galanin-1 receptor
LPS, lipopolysaccharide
NO, nitric oxide
NOS, nitric oxide synthase
iNOS, inducible NOS
EIEC, enteroinvasive E coli
PGE2
prostaglandin E2
PGHS, prostaglandin H synthase
COX-2, cyclooxygenase 2
TLR, toll-like receptor
MHC, major histocompatibility complex
IL, interleukin
PEEC, pathogen elicited epithelial derived chemoattractant
NFκB, nuclear factor κB
TNF-α, tumour necrosis factor α
IκB, inhibitory protein κB
IKK, IκB kinase
NIK, NFκB inducing kinase
MAP, mitogen activated protein
MAPK, MAP kinase
MAPKKK, MAPK kinase kinase
MEK, MAP/ERK kinases
JNK, c-Jun NH2 terminal kinase
AP-1, activating protein 1
cag
cytotoxin associated antigen
REFERENCES
- 1.Ottingler MSL. Clostridium difficile toxin B induces reorganization of actin, vinculin, and talin in cultured cells. Exp Cell Res 1988;174:215–29. [DOI] [PubMed] [Google Scholar]
- 2.Hecht G, Pothoulakis C, LaMont JT, et al. Clostridium difficile toxin A perturbs cytoskeletal structure and tight junction permeability of cultured human intestinal epithelial monolayers. J Clin Invest 1988;82:1516–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hecht G, Koutsouris A, Pothoulakis C, et al. Clostridium difficile toxin B disrupts the barrier function of T84 monolayers. Gastroenterology 1992;102:416–23. [DOI] [PubMed] [Google Scholar]
- 4.Dillon S, Rubin E, Yakubovich M, et al. Involvement of Ras-related Rho proteins in the mechanisms of action of Clostridium difficile Toxin A and Toxin B. Infect Immun 1995;63:1421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Just I, Fritz G, Aktories K, et al. Clostridium difficile toxin B acts on the GTP-binding protein Rho. J Biol Chem 1994;269:10706–12. [PubMed] [Google Scholar]
- 6.Takaishi K, Sasaki T, Kotani H, et al. Regulation of cell-cell adhesion by Rac and Rho small G proteins in MDCK cells. J Cell Biol 1997;139:1047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nusrat A, Giry M, Turner JR, et al. Rho protein regulates tight junctions and perijunctional actin organization in polarized epithelia. Proc Natl Acad Sci USA 1995;92:10629–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Just I, Selzer J, Wilm M, et al. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 1995;375:500–3. [DOI] [PubMed] [Google Scholar]
- 9.Nusrat A, von Eichel-Streiber C, Turner J, et al. Clostridium difficile toxins disrupt epithelial barrier function by altering membrane microdomain localization of tight junction proteins. Infect Immun 2001;69:1329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen M, Pothoulakis C, LaMont T. Protein Kinase C signaling regulates ZO-1 translocation and increased paracellular flux of T84 colonocytes exposed to Clostridium difficile Toxin A. J Biol Chem 2002;277:4247–54. [DOI] [PubMed] [Google Scholar]
- 11.Knutton S, Lloyd D, McNeish A. Adhesion of enteropathogenic Escherichia coli to human intestinal enterocytes and cultured human intestinal mucosa. Infect Immun 1987;55:69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spitz J, Yuhan R, Koutsouris A, et al. Enteropathogenic Escherichia coli adherence to intestinal epithelial monolayers diminishes barrier function. Am J Physiol 1995;268:G374–9. [DOI] [PubMed] [Google Scholar]
- 13.Philpott DJ, McKay DM, Sherman PM, et al. Infection of T84 cells with enteropathogenic Escherichia coli alters barrier and transport. Am J Physiol 1996;270:G634–45. [DOI] [PubMed] [Google Scholar]
- 14.Savkovic SD, Koutsouris A, Hecht G. Attachment of a non-invasive enteric pathogen, enteropathogenic Escherichia coli, to cultured human intestinal epithelial monolayers induces transmigration of neutrophils. Infect Immun 1996;64:4480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuhan R, Koutsouris A, Savkovic S, et al. Enteropathogenic Escherichia coli-induced myosin light chain phosphorylation alters intestinal epithelial permeability. Gastroenterology 1997;113:1873–82. [DOI] [PubMed] [Google Scholar]
- 16.Atisook K, Carlson S, Madara J. Effects of phlorizin and sodium on glucose-elicited alterations of cell junctions in intestinal epithelia. Am J Physiol 1990;258:C77–85. [DOI] [PubMed] [Google Scholar]
- 17.Hecht G, Pestic L, Nikcevic G, et al. Expression of the catalytic domain of myosin light chain kinase increases paracellular permeability. Am J Physiol 1996;271:C1678–84. [DOI] [PubMed] [Google Scholar]
- 18.Turner JR, Rill BK, Carlson SL, et al. Physiological regulation of epithelial tight junctions is associated with myosin light-chain phosphorylation. Am J Physiol 1997;273(4 Pt 1):C1378–85. [DOI] [PubMed] [Google Scholar]
- 19.Furuse M, Hirase T, Itoh M, et al. Occludin: a novel integral membrane protein localizing at tight junctions. J Cell Biol 1993;123:1777–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCarthy K, Skare L, Stankewich M, et al. Occludin is a functional component of the tight junction. J Cell Science 1996;109:2287–98. [DOI] [PubMed] [Google Scholar]
- 21.Wong V. Phosphorylation of occludin correlates with occludin localization and function at the tight junction. Am J Cell Physiol 1997;273:C1859–67. [DOI] [PubMed] [Google Scholar]
- 22.Farshori P, Kachar B. Redistribution and phosphorylation of occludin during opening and resealing of tight junctions in cultured epithelial cells. J Membrane Biol 1999;170:147–56. [DOI] [PubMed] [Google Scholar]
- 23.Simonovic I, Rosenberg J, Koutsouris A, et al. Enteropathogenic E. coli dephosphorylates and dissociates occludin from intestinal epithelial tight junctions. Cell Microbiol 2000;2:305–15. [DOI] [PubMed] [Google Scholar]
- 24.Philpott D, McKay D, Mak W, et al. Signal transduction pathways involved in enterohemorrhagic Escherichia coli-induced alterations in T84 epithelial permeability. Infect Immun 1998;66:1680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koutsouris A, Savkovic S, Hecht G. PKCzeta is activated in intestinal epithelial cells infected by Enteropathogenic E. coli and participates in the alteration of tight junctions. Gastroenterology 2002;122:A56. [Google Scholar]
- 26.Myers L, Shoop D, Stackhouse L, et al. Isolation of enterotoxigenic Bacteroides fragilis from humans with diarrhea. J Clin Microbiol 1987;25:2330–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Myers L, Shoop D, Collins J. Rabbit model to evaluate enterovirulence of Bacteroides fragilis. J Clin Microbiol 1990;28:1658–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moncrief JS, Obiso R, Barroso LA, et al. The enterotoxin of Bacteroides fragilis is a metallaprotease. Infect Immun 1995;63:175–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chambers F, Koshy S, Saidi R, et al. Bacteroides fragilis toxin exhibits polar activity on monolayers of human intestinal epithelial cells (T84 Cells) in vitro. Infect Immun 1997;65:3561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Obiso R, Azghani A, Wilkins T. The Bacteroides fragilis toxin fragilysin disrupts the paracellular barrier of epithelial cells. Infect Immun 1997;65:1431–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koshy S, Montrose M, Sears C. Human intestinal epithelial cells swell and demonstrate actin rearrangement in response to the metalloprotease toxin of Bacteroides fragilis. Infect Immun 1996;64:5022–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reigler M, Lotz M, Sears C, et al. Bacteroides fragilis toxin 2 damages human colonic mucosa in vitro. Gut 1999;44:504–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu S, Lim K, Huang J, et al. Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc Natl Acad Sci USA 1998;95:14979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Booth BA, Boesman-Finkelstein M, Finkelstein RA. Vibrio cholerae hemagglutinin/protease nicks cholera enterotoxin. Infect Immun 1984;45:558–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagamune K, Yamamoto K, Naka A, et al. In vitro proteolytic processing and activation of the recombinant precursor of El Tor cytolysin/hemolysin (pro-HlyA) of Vibrio cholerae by soluble hemagglutinin/protease of V. cholerae, trypsin, and other proteases. Infect Immun 1996;64:4655–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimsey H, Waldor M. Vibrio cholera hemagglutinin/protease inactivates CTXφ. Infect Immun 1998;66:4025–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu Z, Milton D, Nybom P, et al. Vibrio cholerae hemagglutinin/protease (HA/protease) causes morphological changes in cultured epithelial cells and perturbs their paracellular barrier function. Microb Pathog 1996;21:111–23. [DOI] [PubMed] [Google Scholar]
- 38.Wu Z, Nybom P, Magnusson K. Distinct effects of Vibrio cholerae haemagglutinin/protease on the structure and localization of the tight junction-associated proteins occludin and ZO-1. Cell Micriob 2000;2:11–17. [DOI] [PubMed] [Google Scholar]
- 39.Carman R. Clostridium perfringens in spontaneous and antibiotic-associated diarrhoea of man and other animals. Rev Med Microbiol 1997;8(suppl 1):S43. [Google Scholar]
- 40.Abrahao C, Carman R, Hahn H, et al. Similar frequency of detection of Clostridium perfringens enterotoxin and Clostridium difficile toxins in patients with antibiotic-associated diarrhea. Eur J Clin Microbiol Infect Dis 2001;20:676–7. [DOI] [PubMed] [Google Scholar]
- 41.Brett M, Rodhouse J, Donovan T, et al. Detection of Clostridium perfringens and its enterotoxin in cases of sporadic diarrhoea. J Clin Pathol 1992;45:609–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sparks S, Carman R, Sarker M, et al. Genotyping of Enterotoxigenic Clostridium perfringens fecal isolates associated with antibiotic-associated diarrhea and food poisoning in North America. J Clin Microbiol 2001;39:883–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanna P, Mietzner T, McClane B. Localization of the receptor-binding region of Clostridium perfringens enterotoxin utilizing cloned toxin fragments and synthetic peptides. The 30 C-terminal amino acids define a functional binding regions. J Biol Chem 1991;266:11037–43. [PubMed] [Google Scholar]
- 44.Hanna P, Wieckowski E, Mietzner T, et al. Mapping of functional regions of Clostridium perfringens type A enterotoxin. Infect Immun 1992;60:2110–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sherman S, Klein E, McClane B. Clostridium perfringens type A enterotoxin induces tissue damage and fluid accumulation in rabbit ileum. J Diarrhoeal Dis Res 1994;12:200–7. [PubMed] [Google Scholar]
- 46.McDonel J, Duncan C. Regional localization of activity of Clostridium perfringens type A enterotoxin in the rabbit ileum, jejunum, and duodenum. J Infect Dis 1977;136:661–6. [DOI] [PubMed] [Google Scholar]
- 47.McClane B. Clostridium perfringens enterotoxin acts by producing small molecule permeability alterations in plasma membranes. Toxicology 1994;87:43–67. [DOI] [PubMed] [Google Scholar]
- 48.Hardy S, Denmead M, Parekh N, et al. Cationic currents induced by Clostridium perfringens type A enterotoxin in human intestinal CaCO-2 cells. J Med Microbiol 1999;48:235–43. [DOI] [PubMed] [Google Scholar]
- 49.Wieckowski E, Wnek A, McClane B. Evidence that an approximately 50-kDa mammalian plasma membrane protein with receptor-like properties mediates the amphiphilicity of specifically bound Clostridium perfringens entertoxin. J Biol Chem 1994;269:10838–48. [PubMed] [Google Scholar]
- 50.Katahira J, Inoue N, Horiguchi Y, et al. Molecular cloning and functional characterization of the receptor for Clostridium perfringens enterotoxin. J Cell Biol 1997;136:1239–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Katahira J, Sugiyama H, Inoue N, et al. Clostridium perfringens enterotoxin utilizes two structurally related membrane proteins as functional receptors in vivo. J Biol Chem 1997;272:26652–8. [DOI] [PubMed] [Google Scholar]
- 52.Morita K, Furuse M, Fujimoto K, et al. Claudin multigene family encoding four-transmembrane domain protein components of tight junctions strands. Proc Natl Acad Sci USA 1999;96:511–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh U, Van Itallie C, MItic L, et al. CaCo-2 cells treated with Clostridium perfringens enterotoxin form multiple large complex species, one of which contains the tight junction protein occludin. J Biol Chem 2000;275:18407–17. [DOI] [PubMed] [Google Scholar]
- 54.Rahner C, MItic L, McClane B, et al. Clostridium perfringens enterotoxin impairs bile flow in the isolated perfused rat liver and induces fragmentation of tight junction fibrils. Hepatology 1999;30:326A. [Google Scholar]
- 55.Sonoda N, Furuse M, Sasaki T, et al. Clostridium perfringens enterotoxin fragment removes specific claudins from tight junction strands: evidence for direct involvement of claudins in tight junction barrier. J Cell Biol 1999;147:195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barton E, Forrest J, Conolly J, et al. Junction adhesion molecule is a receptor for reovirus. Cell 2001;104:441–51. [DOI] [PubMed] [Google Scholar]
- 57.Cohen C, Shieh J, Pickles R, et al. The coxackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc Natl Acad Sci USA 2001;98:15191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keely S, Barrett K. Regulation of chloride secretion: novel pathways and messengers. Ann NY Acad Sci 2000;915:67–76. [DOI] [PubMed] [Google Scholar]
- 59.Molina N, Peterson J. Cholera toxin-like toxin released by Salmonella species in the presence of mitomycin C. Infect Immun 1980;30:224–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruiz-Palacios G, Torres J, NI, Escamilla E, et al. Cholera-like enterotoxin produced by Campylobacter jejuni. Characterisation and clinical significance. Lancet 1983;2:250–3. [DOI] [PubMed] [Google Scholar]
- 61.Sixma T, Pronk S, Kalk K, et al. Lactose binding to heat-labile enterotoxin revealed by X-ray crystallography. Nature 1992;355:561–4. [DOI] [PubMed] [Google Scholar]
- 62.Wimer-Mackin S, Holmes R, Wolf A, et al. Characterization of receptor-mediated signal transduction by Escherichia coli type IIa heat-labile enterotoxin in the polarized human intestinal cell line T84. Infect Immun 2001;69:7205–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Field M, Rao M, Chang E. Intestinal electrolyte transport and diarrheal disease (2). N Engl J Med 1990;321:879–83. [DOI] [PubMed] [Google Scholar]
- 64.Gabriel S, Brigman K, Koller B, et al. Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 1995;266:107–9. [DOI] [PubMed] [Google Scholar]
- 65.Turvill J, Mourad F, Farthing M. Crucial role for 5-HT in cholera toxin but not Escherichia coli heat-labile enterotoxin-intestinal secretion in rats. Gastroenterology 1998;115:883–90. [DOI] [PubMed] [Google Scholar]
- 66.Vaandrager A, Tilly B, Smolenski A, et al. cGMP stimulation of cystic fibrosis transmembrane conductance regulator Cl- channels co-expressed with cGMP-dependent protein kinase type II but not type Iβ. J Biol Chem 1997;272:4195–200. [DOI] [PubMed] [Google Scholar]
- 67.Currie MG, Fok KF, Kato J, et al. Guanylin: an endogenous activator of intestinal guanylate cyclase. Proc Natl Acad Sci USA 1992;89:947–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamra FK, Forte LR, Eber SL, et al. Uroguanylin: structure and activity of a second endogenous peptide that stimulates intestinal guanylate cyclase. Proc Natl Acad Sci USA 1993;90:10464–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulz S, Green C, Yuen P, et al. Guanylyl cylase is a heat-stable enterotoxin receptor. Cell 1990;63:941–8. [DOI] [PubMed] [Google Scholar]
- 70.Savarino S, Fasano A, Robertson D, et al. Enteroaggregative Escherichia coli elaborate a heat-stable enterotoxin demonstrable in an in vitro rabbit intestinal model. J Clin Invest 1991;87:1450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Savarino S, Fasano A, Watson J, et al. Enteroaggregative Escherichia coli heat-stable Enterotoxin 1 represents another subfamily of E. coli heat-stable toxin. Proc Natl Acad Sci USA 1993;90:3093–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.de Sousa P, Dubreuil J. Distribution and expression of the astA gene (EAST1 toxin) in Escherichia coli and Salmonella. J Med Microbiol 2001;291:15–20. [DOI] [PubMed] [Google Scholar]
- 73.Savarino S, McVeigh A, Watson J, et al. Enteroaggregative Escherichia coli heat-stable enterotoxin is not restricted to enteroaggregative E. coli. J Infect Dis 1996;173:1019–22. [DOI] [PubMed] [Google Scholar]
- 74.Rao MC, Guandalini S, Laird WJ, et al. Effects of heat-stable enterotoxin of Yersinia enterocolitica on ion transport and cyclic guanosine 3′,5′-monophosphate metabolism in rabbit ileum. Infect Immun 1979;26:875–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Takeda T, Peina Y, Ogawa A, et al. Detection of heat-stable enterotoxin in a cholera toxin gene-positive strain of Vibro cholerai O1. FEMS Microbiol Lett 1991;64:23–7. [DOI] [PubMed] [Google Scholar]
- 76.Guarino A, Guandalini S, Alessio M, et al. Characteristics and mechanism of action of a heat-stable enterotoxin produced by Klebsiella pneumoniae from infants with secretory diarrhea. Pediatr Res 1989;25:514–18. [DOI] [PubMed] [Google Scholar]
- 77.Bolton J, Field M. Ca ionophore-stimulated ion secretion in rabbit ileal mucosa: relation to actions of cyclic 3′, 5′-AMP and carbamylcholine. J Membr Biol 1977;35:159–73. [DOI] [PubMed] [Google Scholar]
- 78.Donowitz M, Asarkof N, Pike G. Calcium dependence of serotonin-induced changes in rabbit ileal electrolyte transport. J Clin Inves 1980;66:341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cuthbert A, Margolius H. Kinins stimulate net chloride secretion by the rat colon. Br J Pharm 1982;75:587–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.McEwan GT, Hirst BH, Simmons NL. Carbachol stimulates Cl-secretion via activation of two distinct apical Cl-pathways in cultured human T84 intestinal epithelial monolayers. Biochim Biophys Acta 1994;1220:241–7. [DOI] [PubMed] [Google Scholar]
- 81.Gruber AD, Elble RC, Ji HL, et al. Genomic cloning, molecular characterization, and functional analysis of Human CLCA1, the first human member of the family of Ca2+-activated Cl− channel proteins. Genomics 1998;54:200–14. [DOI] [PubMed] [Google Scholar]
- 82.Fuller C, Ismailov I, Keeton D, et al. Phosphorylation and activation of a bovine tracheal anion channel by Ca2+/calmodulin-dependent protein kinase II. J Biol Chem 1994;269:26642–50. [PubMed] [Google Scholar]
- 83.Raimondi F, Kao J, Fiorentini C, et al. Enterotoxicity and cytotoxicity of Vibrio parahaemolyticus thermostable director Hemolysin in In Vitro systems. Infect Immun 2000;68:3180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Takahashi A, Sato Y, Shiomi Y, et al. Mechanisms of chloride secretion induced by thermostable direct haemolysin of Vibrio parahaemolyticus in human colonic tissue and a human intestinal epithelial cell line. J Med Microbiol 2000;49:801–10. [DOI] [PubMed] [Google Scholar]
- 85.Takahashi A, Kenjyo N, Imura K, et al. Cl-Secretion in colonic epithelial cells induced by the Vibrio parahaemolyticus hemolytic toxin related to thermostable direct hemolysin. Infect Immun 2000;68:5435–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lundgren O, Svensson L. Pathogenesis of rotavirus diarrhea. Microbes Infect 2001;3:1145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ball J, Tian P, Zeng C, et al. Age-dependent diarrhea induced by a rotaviral nonstructural glycoprotein. Science 1996;272:101–4. [DOI] [PubMed] [Google Scholar]
- 88.Tian P, Hu Y, Schilling W, et al. The nonstructural glycoprotein of rotavirus affects intracellular calcium levels. J Virol 1994;68:251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dong Y, Zeng C, Ball J, et al. The rotavirus enterotoxin NSP4 mobilizes intracellular calcium in human intestinal cells by stimulating phospholipase C-mediated inositol 1,4,5-trisphosphate production. Proc Natl Acad Sci USA 1997;94:3960–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoyle C, Burnstock G. Galanin-like immunoreactivity in enteric neurons of the human colon. J Anatomy 1989;166:23–33. [PMC free article] [PubMed] [Google Scholar]
- 91.Katsoulis S, Clemens A, Morys-Wortmann C, et al. Human galanin modulates human colonic motility in vitro. Characterization of structural requirements. Scand J Gastroenterol 1996;31:446–51. [DOI] [PubMed] [Google Scholar]
- 92.Benya RV, Marrero JA, Ostrovkiy D, et al. Human colonic epithelial cells express galanin-1 receptors which when activated cause Cl-secretion. Am J Physiol 1999;276:G64–72. [DOI] [PubMed] [Google Scholar]
- 93.Lorimer DD, Benya RV. Cloning and quantification of human galanin-1 receptor expression by mucosal cells lining the gastrointestinal tract. Biochem Biophys Res Commun 1996;222:379–85. [DOI] [PubMed] [Google Scholar]
- 94.Hecht G, Marrero JA, Danilkovich A, et al. Pathogenic Escherichia coli increase Cl-secretion from intestinal epithelia by upregulating galanin-1 receptor expression. J Clin Invest 1999;104:253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Marrero JA, Matkowskyj KA, Yung K, et al. Dextran sulfate sodium-induced murine colitis activates NF-κB and increases galanin-1 receptor expression. Am J Physiol Gastrointest Liver Physiol 2000;278:G797–804. [DOI] [PubMed] [Google Scholar]
- 96.Matkowskyj KA, Danilkovich A, Marrero JA, et al. Galanin-1 receptor up-regulation mediates the excess colonic fluid production caused by infection with enteric pathogens. Nat Med 2000;6:1048–51. [DOI] [PubMed] [Google Scholar]
- 97.Shaw RD, Hempson SJ, Matkowskyj KA, et al. Galanin-1 receptor expression contributes to Rotavirus Diarrhea. Gastroenterology 2002;122:A76. [Google Scholar]
- 98.Resta-Lenert S, Barrett K. Enteroinvasive bacteria alter barrier and transport properties of human intestinal epithelium: Role of iNOS and COX-2. Gastroenterology 2002;122:1070–87. [DOI] [PubMed] [Google Scholar]
- 99.Fang F. Mechanisms of nitric oxide-related antimicrobial activity. J Clin Invest 1997;99:2818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.De Groote M, Fang F. NO inhibitions: antimicrobial properties of nitric oxide. Clin Infect Dis 1995;21(suppl 2):S162–5. [DOI] [PubMed] [Google Scholar]
- 101.Alican I, Kubes P. A critical role for nitric oxide in intestinal barrier function and dysfunction. Am J Physiol 1996;270(2 Pt 1):G225–37. [DOI] [PubMed] [Google Scholar]
- 102.Nathan C, Xie Q. Nitric oxide synthases: Roles, tolls and controls. Cell 1994;78:915. [DOI] [PubMed] [Google Scholar]
- 103.Rolfe V, Milla P. Nitric oxide stimulates cyclic guanosine monophosphate production and electrogenic secretion in Caco-2 colonocytes. Clin Sci 1999;96:165–70. [PubMed] [Google Scholar]
- 104.Tepperman B, Brown J, Korolkiewicz R, et al. Nitric oxide synthase activity, viability and cyclic GMP levels in rat colonic epithelial cells: effect of endotoxin challenge. Am Soc Pharm Exp Ther 1994;271:1477–82. [PubMed] [Google Scholar]
- 105.Witthoft T, Eckmann L, Kim J, et al. Enteroinvasive bacteria directly activate expression of iNOS and NO production in human colon epithelial cells. Am J Physiol 1998;275:G564–71. [DOI] [PubMed] [Google Scholar]
- 106.Islam D, Veress B, Bardhan P, et al. In situ characterization of inflammatory responses in the rectal mucosae of patients with Shigellosis. Infect Immun 1997;65:739–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Salzman A, Eaves-Pyles T, Linn S, et al. Bacterial induction of inducible nitric oxide synthase in cultured human intestinal epithelial cells. Gastroenterology 1998;114:93–102. [DOI] [PubMed] [Google Scholar]
- 108.Weymer A, Huott P, Liu W, et al. Chloride secretory mechanism induced by prostaglandin E1 in a colonic epithelial cell line. J Clin Inves 1985;76:1828–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Smith W, Dewitt D. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol 1996;62:167–215. [DOI] [PubMed] [Google Scholar]
- 110.Eckmann L, Stenson W, Savidge T, et al. Role of intestinal epithelial cells in the host secretory response to infection by invasive bacteria. J Clin Invest 1997;100:296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Laurent F, Kagnoff M, Savidge T, et al. Human intestinal epithelial cells repond to Cryptosporidium parvum infection with increased prostaglandin H synthase 2 expression and prostaglandin E2 and F2alpha production. Infect Immun 1998;66:1787–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pomorski T, Meyer T, Naumann M. Helicobacter pylori-induced prostaglandin E2 syntheseis involves activation of cytosolic phospholipase A2 in epithelial cells. J Biol Chem 2001;276:804–10. [DOI] [PubMed] [Google Scholar]
- 113.Stenson W, Zhang A, Riehl T, et al. Ambeic infection in the human colon induces cyclooxygenase-2. Infect Immun 2001;69:3382–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Caamano J, Hunter CA. NF-kappaB family of transcription factors: central regulators of innate and adaptive immune functions. Clin Microbiol Rev 2002;15:414–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.McCormick BA, Hofman PM, Kim J, et al. Surface attachment of Salmonella typhimurium to intestinal epithelia imprints the subepithelial matrix with gradients chemotactic for neutrophils. J Cell Biol 1995;131(6 Pt 1):1599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.McCormick BA, Parkos CA, Colgan SP, et al. Apical secretion of a pathogen-elicited epithelial chemoattractant activity in response to surface colonization of intestinal epithelia by Salmonella typhimurium. J Immunol 1998;160:455–66. [PubMed] [Google Scholar]
- 117.Madara JL, Patapoff TW, Gillece-Castro B, et al. 5′-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers (see comments). J Clin Invest 1993;91:2320–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Strohmeier GR, Reppert SM, Lencer WI, et al. The A2b adenosine receptor mediates cAMP responses to adenosine receptor agonists in human intestinal epithelia. J Biol Chem 1995;270:2387–94. [DOI] [PubMed] [Google Scholar]
- 119.Li ZW, Chu W, Hu Y, et al. The IKKbeta subunit of IkappaB kinase (IKK) is essential for nuclear factor kappaB activation and prevention of apoptosis. J Exp Med 1999;189:1839–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Force WR, Glass AA, Benedict CA, et al. Discrete signaling regions in the lymphotoxin-beta receptor for tumor necrosis factor receptor-associated factor binding, subcellular localization, and activation of cell death and NF-kappaB pathways. J Biol Chem 2000;275:11121–9. [DOI] [PubMed] [Google Scholar]
- 121.Daig R, Andus T, Aschenbrenner E, et al. Increased interleukin 8 expression in the colon mucosa of patients with inflammatory bowel disease. Gut 1996;38:216–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mazzucchelli L, Hauser C, Zgraggen K, et al. Expression of interleukin-8 gene in inflammatory bowel disease is related to the histological grade of active inflammation. Am J Pathol 1994;144:997–1007. [PMC free article] [PubMed] [Google Scholar]
- 123.Hosokawa T, Kusugami K, Ina K, et al. Interleukin-6 and soluble interleukin-6 receptor in the colonic mucosa of inflammatory bowel disease. J Gastroenterol Hepatol 1999;14:987–96. [DOI] [PubMed] [Google Scholar]
- 124.Wang D, Westerheide SD, Hanson JL, et al. Tumor necrosis factor alpha-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem 2000;275:32592–7. [DOI] [PubMed] [Google Scholar]
- 125.Bird TA, Schooley K, Dower SK, et al. Activation of nuclear transcription factor NF-kappaB by interleukin-1 is accompanied by casein kinase II-mediated phosphorylation of the p65 subunit. J Biol Chem 1997;272:32606–12. [DOI] [PubMed] [Google Scholar]
- 126.Zhong H, Voll RE, Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1998;1:661–71. [DOI] [PubMed] [Google Scholar]
- 127.Anrather J, Csizmadia V, Soares MP, et al. Regulation of NF-kappaB RelA phosphorylation and transcriptional activity by p21(ras) and protein kinase Czeta in primary endothelial cells. J Biol Chem 1999;274:13594–603. [DOI] [PubMed] [Google Scholar]
- 128.Martin AG, San-Antonio B, Fresno M. Regulation of nuclear factor kappa B transactivation. Implication of phosphatidylinositol 3-kinase and protein kinase C zeta in c-Rel activation by tumor necrosis factor alpha. J Biol Chem 2001;276:15840–9. [DOI] [PubMed] [Google Scholar]
- 129.Savkovic S, Koutsouris A, Hecht G. Atypical PKC zeta regulates EPEC-induced inflammation through interaction with IkappaB kinase. Gastroenterology 2002;122:A76. [Google Scholar]
- 130.Lee JC, Laydon JT, McDonnell PC, et al. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 1994;372:739–46. [DOI] [PubMed] [Google Scholar]
- 131.Lee JC, Young PR. Role of CSB/p38/RK stress response kinase in LPS and cytokine signaling mechanisms. J Leukoc Biol 1996;59:152–7. [DOI] [PubMed] [Google Scholar]
- 132.Schlessinger J. SH2/SH3 signaling proteins. Curr Opin Genet Dev 1994;4:25–30. [DOI] [PubMed] [Google Scholar]
- 133.Ueda Y, Hirai S, Osada S, et al. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J Biol Chem 1996;271:23512–9. [DOI] [PubMed] [Google Scholar]
- 134.Berra E, Diaz-Meco MT, Dominguez I, et al. Protein kinase C zeta isoform is critical for mitogenic signal transduction. Cell 1993;74:555–63. [DOI] [PubMed] [Google Scholar]
- 135.Berra E, Diaz-Meco MT, Lozano J, et al. Evidence for a role of MEK and MAPK during signal transduction by protein kinase C zeta. Embo J 1995;14:6157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Monick MM, Carter AB, Gudmundsson G, et al. A phosphatidylcholine-specific phospholipase C regulates activation of p42/44 mitogen-activated protein kinases in lipopolysaccharide-stimulated human alveolar macrophages. J Immunol 1999;162:3005–12. [PubMed] [Google Scholar]
- 137.Monick MM, Carter AB, Flaherty DM, et al. Protein kinase C zeta plays a central role in activation of the p42/44 mitogen-activated protein kinase by endotoxin in alveolar macrophages. J Immunol 2000;165:4632–9. [DOI] [PubMed] [Google Scholar]
- 138.Sajan MP, Standaert ML, Bandyopadhyay G, et al. Protein kinase C-zeta and phosphoinositide-dependent protein kinase-1 are required for insulin-induced activation of ERK in rat adipocytes. J Biol Chem 1999;274:30495–500. [DOI] [PubMed] [Google Scholar]
- 139.Mercurio F, Zhu H, Murray BW, et al. IKK-1 and IKK-2: cytokine-activated IkappaB kinases essential for NF-kappaB activation. Science 1997;278:860–6. [DOI] [PubMed] [Google Scholar]
- 140.Nakano H, Shindo M, Sakon S, et al. Differential regulation of I kappa B kinase alpha and beta by two upstream kinases, NF-kappa B-inducing kinase and mitogen-activated protein kinase/ERK kinase kinase-1. Proc Natl Acad Sci USA 1998;95:3537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Stabel S, Parker PJ. Protein kinase C. Pharmacol Ther 1991;51:71–95. [DOI] [PubMed] [Google Scholar]
- 142.Hug H, Sarre TF. Protein kinase C isoenzymes: divergence in signal transduction? Biochem J 1993;291(Pt 2):329–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Dekker LV, Parker PJ. Protein kinase C—a question of specificity. Trends Biochem Sci 1994;19:73–7. [DOI] [PubMed] [Google Scholar]
- 144.Diaz-Meco MT, Municio MM, Frutos S, et al. The product of par-4, a gene induced during apoptosis, interacts selectively with the atypical isoforms of protein kinase C. Cell 1996;86:777–86. [DOI] [PubMed] [Google Scholar]
- 145.Moscat J, Diaz-Meco MT. The atypical protein kinase Cs. Functional specificity mediated by specific protein adapters. EMBO Rep 2000;1:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Akimoto K, Takahashi R, Moriya S, et al. EGF or PDGF receptors activate atypical PKClambda through phosphatidylinositol 3-kinase. EMBO J 1996;15:788–98. [PMC free article] [PubMed] [Google Scholar]
- 147.Bjorkoy G, Overvatn A, Diaz-Meco MT, et al. Evidence for a bifurcation of the mitogenic signaling pathway activated by Ras and phosphatidylcholine-hydrolyzing phospholipase C. J Biol Chem 1995;270:21299–306. [DOI] [PubMed] [Google Scholar]
- 148.Bjorkoy G, Perander M, Overvatn A, et al. Reversion of Ras- and phosphatidylcholine-hydrolyzing phospholipase C-mediated transformation of NIH 3T3 cells by a dominant interfering mutant of protein kinase C lambda is accompanied by the loss of constitutive nuclear mitogen-activated protein kinase/extracellular signal-regulated kinase activity. J Biol Chem 1997;272:11557–65. [DOI] [PubMed] [Google Scholar]
- 149.Diaz-Meco MT, Berra E, Municio MM, et al. A dominant negative protein kinase C zeta subspecies blocks NF-kappa B activation. Mol Cell Biol 1993;13:4770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Diaz-Meco MT, Lozano J, Municio MM, et al. Evidence for the in vitro and in vivo interaction of Ras with protein kinase C zeta. J Biol Chem 1994;269:31706–10. [PubMed] [Google Scholar]
- 151.Diaz-Meco MT, Dominguez I, Sanz L, et al. PKC zeta induces phosphorylation and inactivation of I kappa B-alpha in vitro. EMBO J 1994;13:2842–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Schonwasser DC, Marais RM, Marshall CJ, et al. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Mol Cell Biol 1998;18:790–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rahman A, Anwar KN, Malik AB. Protein kinase C-zeta mediates TNF-alpha-induced ICAM-1 gene transcription in endothelial cells. Am J Physiol Cell Physiol 2000;279:C906–14. [DOI] [PubMed] [Google Scholar]
- 154.Folgueira L, McElhinny JA, Bren GD, et al. Protein kinase C-zeta mediates NF-kappa B activation in human immunodeficiency virus-infected monocytes. J Virol 1996;70:223–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sontag E, Sontag JM, Garcia A. Protein phosphatase 2A is a critical regulator of protein kinase C zeta signaling targeted by SV40 small t to promote cell growth and NF-kappaB activation. EMBO J 1997;16:5662–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lozano J, Berra E, Municio MM, et al. Protein kinase C zeta isoform is critical for kappa B-dependent promoter activation by sphingomyelinase. J Biol Chem 1994;269:19200–2. [PubMed] [Google Scholar]
- 157.Jung HC, Eckmann L, Yang SK, et al. A distinct array of proinflammatory cytokines is expressed in human colon epithelial cells in response to bacterial invasion. J Clin Invest 1995;95:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kampik D, Schulte R, Autenrieth IB. Yersinia enterocolitica invasin protein triggers differential production of interleukin-1, interleukin-8, monocyte chemoattractant protein 1, granulocyte-macrophage colony-stimulating factor, and tumor necrosis factor alpha in epithelial cells: implications for understanding the early cytokine network in Yersinia infections. Infect Immun 2000;68:2484–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Eaves-Pyles T, Szabo C, Salzman AL. Bacterial invasion is not required for activation of NF-kappaB in enterocytes. Infect Immun 1999;67:800–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Gewirtz AT, Simon PO Jr, Schmitt CK, et al. Salmonella typhimurium translocates flagellin across intestinal epithelia, inducing a proinflammatory response. J Clin Invest 2001;107:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.McCormick BA, Colgan SP, Delp-Archer C, et al. Salmonella typhimurium attachment to human intestinal epithelial monolayers: transcellular signalling to subepithelial neutrophils. J Cell Biol 1993;123:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sitaraman SV, Merlin D, Wang L, et al. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J Clin Invest 2001;107:861–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Reed KA, Hobert ME, Kolenda CE, et al. The Salmonella typhimurium flagellar basal body protein FliE is required for flagellin production and to induce a proinflammatory response in epithelial cells. J Biol Chem 2002;277:13346–53. [DOI] [PubMed] [Google Scholar]
- 164.Eaves-Pyles T, Murthy K, Liaudet L, et al. Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: I kappa B alpha degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J Immunol 2001;166:1248–60. [DOI] [PubMed] [Google Scholar]
- 165.Gewirtz AT, Navas TA, Lyons S, et al. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol 2001;167:1882–5. [DOI] [PubMed] [Google Scholar]
- 166.Gewirtz AT, Siber AM, Madara JL, et al. Orchestration of neutrophil movement by intestinal epithelial cells in response to Salmonella typhimurium can be uncoupled from bacterial internalization. Infect Immun 1999;67:608–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gewirtz A, Rao A, Simon PJ, et al. Salmonella typhimurium induces epithelial IL-8 expression via Ca2+-mediated activation of the NF-kB pathway. J Clin Invest 2000;105:79–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hobbie S, Chen LM, Davis RJ, et al. Involvement of mitogen-activated protein kinase pathways in the nuclear responses and cytokine production induced by Salmonella typhimurium in cultured intestinal epithelial cells. J Immunol 1997;159:5550–9. [PubMed] [Google Scholar]
- 169.Mynott T, Crossett B, Prathalingam S. Proteolytic inhibition of Salmonella enterica serovar typhimurium-induced activation of the mitogen-activated protein kinases ERK and JNK in cultured human intestinal cells. Infect Immun 2002;70:86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Galdiero M, Vitiello M, Sanzari E, et al. Porins from Salmonella enterica serovar Typhimurium activate the transcription factors activating protein 1 and NF-kappaB through the Raf-1-mitogen-activated protein kinase cascade. Infect Immun 2002;70:558–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lee CA, Silva M, Siber AM, et al. A secreted Salmonella protein induces a proinflammatory response in epithelial cells, which promotes neutrophil migration. Proc Natl Acad Sci USA 2000;97:12283–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Criss AK, Silva M, Casanova JE, et al. Regulation of Salmonella-induced neutrophil transmigration by epithelial ADP-ribosylation factor 6. J Biol Chem 2001;276:48431–9. [DOI] [PubMed] [Google Scholar]
- 173.McCormick BA, Miller S, Carnes D, et al. Transepithelial signaling to neutrophils by Salmonellae: a novel virulence mechanism for gastroenteritis. Infect Immun 1995;63:2302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Kenny B, Finlay BB. Intimin-dependent binding of enteropathogenic Escherichia coli to host cells triggers novel signaling events, including tyrosine phosphorylation of phospholipase C-gamma1. Infect Immun 1997;65:2528–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Savkovic S, Koutsouris A, Hecht G. Activation of NF-κB in intestinal epithelial cells by enteropathogenic Escherichia coli. Am J Physiol 1997;273:C1160–7. [DOI] [PubMed] [Google Scholar]
- 176.Savkovic SD, Ramaswamy A, Koutsouris A, et al. EPEC-activated ERK1/2 participate in inflammatory response but not tight junction barrier disruption. Am J Physiol Gastrointest Liver Physiol 2001;281:G890–8. [DOI] [PubMed] [Google Scholar]
- 177.Czerucka D, Dahan S, Mograbi B, et al. Implication of mitogen-activated protein kinases in T84 cell responses to enteropathogenic Escherichia coli infection. Infect Immun 2001;69:1298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Rosenshine I, Donnenberg MS, Kaper JB, et al. Signal transduction between enteropathogenic Escherichia coli (EPEC) and epithelial cells: EPEC induces tyrosine phosphorylation of host cell proteins to initiate cytoskeletal rearrangement and bacterial uptake. EMBO J 1992;11:3551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Celli J, Olivier M, Finlay BB. Enteropathogenic Escherichia coli mediates antiphagocytosis through the inhibition of PI 3-kinase-dependent pathways. EMBO J 2001;20:1245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Crane JK, Oh JS. Activation of host cell protein kinase C by enteropathogenic Escherichia coli. Infect Immun 1997;65:3277–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Branka J, Vallettte G, Jarry A, et al. Early functional effects of Clostridium difficile toxin A on human colonocytes. Gastroenterology 1997;112:1887–94. [DOI] [PubMed] [Google Scholar]
- 182.Mahida YR, Makh S, Hyde S, et al. Effect of Clostridium difficile toxin A on human intestinal epithelial cells: induction of interleukin 8 production and apoptosis after cell detachment. Gut 1996;38:337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jefferson KK, Smith MF Jr, Bobak DA. Roles of intracellular calcium and NF-kappa B in the Clostridium difficile toxin A-induced up-regulation and secretion of IL-8 from human monocytes. J Immunol 1999;163:5183–91. [PubMed] [Google Scholar]
- 184.Mounier J, Vasselon T, Hellio R, et al. Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infect Immun 1992;60:237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Raqib R, Lindberg AA, Wretlind B, et al. Persistence of local cytokine production in Shigellosis in acute and convalescent stages. Infect Immun 1995;63:289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Sansonetti PJ, Arondel J, Huerre M, et al. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun 1999;67:1471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Philpott DJ, Yamaoka S, Israel A, et al. Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol 2000;165:903–14. [DOI] [PubMed] [Google Scholar]
- 188.Kohler H, Rodrigues SP, McCormick BA. Shigella flexneri interactions with the basolateral membrane domain of polarized model intestinal epithelium: role of lipopolysaccharide in cell invasion and in activation of the mitogen-activated protein kinase ERK. Infect Immun 2002;70:1150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Giarardin SE, Tournebize R, Mavris M, et al. CARD4/Nod1 mediates NF-KappaB and JNK activation by invasive Shigella flexneri. EMBO J 2001;2:736–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Parsonnet J, Friedman GD, Vandersteen DP, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991;325:1127–31. [DOI] [PubMed] [Google Scholar]
- 191.Parsonnet J, Hansen S, Rodriguez L, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med 1994;330:1267–71. [DOI] [PubMed] [Google Scholar]
- 192.Covacci A, Censini S, Bugnoli M, et al. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci USA 1993;90:5791–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Weel JF, van der Hulst RW, Gerrits Y, et al. The interrelationship between cytotoxin-associated gene A, vacuolating cytotoxin, and Helicobacter pylori-related diseases. J Infect Dis 1996;173:1171–5. [DOI] [PubMed] [Google Scholar]
- 194.Peek RM Jr, Miller GG, Tham KT, et al. Heightened inflammatory response and cytokine expression in vivo to cagA+ Helicobacter pylori strains. Lab Invest 1995;73:760–70. [PubMed] [Google Scholar]
- 195.Crabtree JE, Farmery SM, Lindley IJ, et al. CagA/cytotoxic strains of Helicobacter pylori and interleukin-8 in gastric epithelial cell lines. J Clin Pathol 1994;47:945–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Crabtree JE, Wyatt JI, Trejdosiewicz LK, et al. Interleukin-8 expression in Helicobacter pylori infected, normal, and neoplastic gastroduodenal mucosa. J Clin Pathol 1994;47:61–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Sharma SA, Tummuru MK, Blaser MJ, et al. Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol 1998;160:2401–7. [PubMed] [Google Scholar]
- 198.Naumann M, Wessler S, Bartsch C, et al. Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. J Biol Chem 1999;274:31655–62. [DOI] [PubMed] [Google Scholar]
- 199.Glocker E, Lange C, Covacci A, et al. Proteins encoded by the cag pathogenicity island of Helicobacter pylori are required for NF-kappaB activation. Infect Immun 1998;66:2346–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Maeda S, Yoshida H, Ogura K, et al. H. pylori activates NF-kappaB through a signaling pathway involving IkappaB kinases, NF-kappaB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology 2000;119:97–108. [DOI] [PubMed] [Google Scholar]
- 201.Keates S, Keates AC, Warny M, et al. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol 1999;163:5552–9. [PubMed] [Google Scholar]
- 202.Rieder G, Wolfgang E, Hatz R, et al. Comparison of CXC chemokines ENA-78 and Interleukin-8 expression in Helicobacter pylori-associated Gastritis. Infect Immun 2001;69:81–8. [DOI] [PMC free article] [PubMed] [Google Scholar]