

Abstract
Backgrounds: The Raf/MEK/ERK (mitogen activated protein kinase—MAPK) signal transduction cascade is an important mediator of a number of cellular fates, including growth, proliferation, and survival. The BRAF gene, one of the human isoforms of RAF, is activated by oncogenic Ras, leading to cooperative effects in cells responding to growth factor signals.
Aims: The aim of this study was to elucidate a possible function of BRAF in liver tumours.
Methods: Mutations of BRAF and KRAS were evaluated in 25 hepatocellular carcinomas (HCC) and in 69 cholangiocarcinomas (CC) by direct DNA sequencing analyses after microdissection. The presence of active intermediates of the MAPK pathway was assessed immunohistochemically. The results obtained were correlated with histopathological variables and patient survival.
Results: Activating BRAF missense mutations were identified in 15/69 CC (22%) and in one case of tumour surrounding liver. KRAS mutations were found in 31 of 69 (45%) CC examined and in two cases of tumour surrounding non-neoplastic liver tissue. In HCC, neither BRAF nor KRAS mutations were detected. All 31 CC with KRAS mutations had an intact BRAF gene. We failed to observe a correlation between BRAF or KRAS mutations and histopathological factors or prognosis of patients.
Conclusions: Our data indicate that BRAF gene mutations are a relatively common event in CC but not in HCC. Disruption of the Raf/MEK/ERK (MAPK) kinase pathway, either by RAS or BRAF mutation, was detected in approximately 62% of all CC and is therefore one of the most frequent defects in cholangiocellular carcinogenesis.
Keywords: cholangiocarcinoma, hepatocellular carcinoma, BRAF, KRAS, histopathology, prognosis
Hepatocellular carcinoma (HCC) is the most frequent malignant primary hepatic neoplasm, followed by intrahepatic cholangiocarcinoma (CC), originating from biliary epithelia or cholangiocytes.1 Whereas many aetiological factors have been characterised for HCC (for example, cirrhosis, hepatitis), the cause of CC remains speculative.2,3 To date, the cellular and molecular mechanisms leading to oncogenesis of hepatocytes or cholangiocytes remain unclear. Increasing evidence exists that carcinogenesis must be understood in terms of accumulation of mutations in regulatory genes, including activation of oncogenes and inactivation or loss of tumour suppressor genes.3,4 Since the discovery of the role of RAS oncogenes in tumorigenesis, we have witnessed an explosion of research in the signal transduction area.4,5 A key RAS effector pathway involves the kinase cascade RAF/MEK/ERK (MEK: MAP/ERK kinase; ERK: extracellular signal related kinase). In the quest to understand how RAS transmits extracellular growth signals, the mitogen activated protein kinase (MAPK) pathway has emerged as an important route between membrane bound RAS and the nucleus.6–8
Signalling through the MAPK cascade is transduced by GTP loading of RAS leading to activation of RAF kinase. In mammalian cells, there are three isoforms of RAF: A-RAF, B-RAF, and C-RAF.9 Although all three of the RAF isoforms share a common function with respect to MEK phosphorylation, studies have shown that these proteins might be differentially activated by oncogenic RAS.9–12 We and others have recently described that activating KRAS mutations may play a role in the carcinogenesis of CC of the liver.13,14 Recently, mutations of BRAF have been described in approximately 15% of all human cancers, especially in malignant melanomas.15 In the present study, we analysed the status of the BRAF gene together with K-RAS to elucidate a possible role of these genes in hepatic malignancies.
MATERIALS AND METHODS
Patients and tissue samples
Sixty nine patients with CC and 25 with HCC undergoing partial hepatectomy (segmental or lobar resection) between 1994 and 2000 were included in this retrospective study. No patient received preoperative or adjuvant chemo- or radiotherapy. All patients underwent surgery for curative intent (R0 resections). Patients who received orthotopic liver transplantation were excluded from the study.
Tumour typing and staging were performed using WHO16 and UICC (2002)17 criteria. In all cases, slides prepared from four different paraffin blocks of tissue, sampled from different tumour areas, were examined.
Pathohistological data are summarised in table 1 ▶.
Table 1.
Patients and pathohistological data
| No of patients | One year survival rate (%) (95% CI) | Median survival time (days) (95% CI) | p Value | |
| Hepatocellular carcinoma (n=25) | ||||
| Stage I† | 4 (16%) | 49 (23–89) | 41 (0, 1098) | |
| Stage II | 9 (36%) | 61 (39–79) | 780 (0, 2820) | p<0.05* |
| Stage IIIA | 10 (40%) | 48 (36–62) | 331 (115, 563) | |
| Stage IIIB | 1 (4%) | 25 (2–49) | 110 (23, 189) | |
| Stage IIIC | 1 (4%) | 21 (8–41) | 98 (21, 191) | |
| Stage IV | — | — | — | |
| Cholangiocarcinoma (n=69) | ||||
| Stage I | 12 (17%) | 100 | 12 patients alive | |
| Stage II | 18 (26%) | 100 | 18 patients alive | |
| Stage IIIA | 20 (29%) | 61 (41–90) | 420 (126, 530) | |
| Stage IIIB | 13 (19%) | 50 (49–98) | 361 (70, 280) | |
| Stage IIIC | 6 (9%) | 39 (10–81) | 201 (19, 297) | |
| Stage IV | — | — | — | |
| Cholangiocarcinoma | ||||
| Braf mutation | ||||
| Present | 15 (22%) | 80 (49–159) | 397 (212, 496) | |
| Absent | 29 (50%) | 71 (41–191) | 364 (19, 512) | NS |
| KRAS mutation | ||||
| Present | 31 (45%) | 81 (51–184) | 399 (178, 512) | |
| Absent | 38 (55%) | 69 (10–174) | 349 (189, 719) | NS |
DNA samples
For each liver tumour sample, the histopathological lesions of interest were first identified on routinely stained rapid frozen sections. Sections (12 μm) were cut from frozen tissue blocks and mounted on glass slides with a thickness of 0.17 mm (very thin glass slides are needed to prevent laser energy from being dispersed before reaching the section of tissue). An ultraviolet laser microscope system was then used to isolate particular cell populations (UV laser microbeam; PALM, Bernried, Germany) (fig 1 ▶). The pulsed UV laser of high beam quality (nitrogen laser, wavelength 337 nm, maximum frequency 20 pulses/s, pulse duration 3 ns) was combined with an inverse microscope and focused through an objective of high numerical aperture into the tissue plane. Beam spot diameter measured approximately 0.3–0.5 μm. Because of the extremely high energy density within the focal point (laser energy at object plane approximately 5 μJ), all biological material is completely destroyed. Using the UV laser beam at a high repetition rate (approximately 20 pulses/s), a circle was cut around the target cells (fig 1A, B ▶). This resulted in complete separation of the target population from neighbouring tissues (fig 1B ▶). The approximate number of cells was estimated to be at least 1000 per sample for polymerase chain reaction (PCR) analysis. In case of non-neoplastic liver samples, microdissection was also performed, especially to select areas without necrosis and severe cholestasis. After microdissection, tissue samples were put into Eppendorf tubes and incubated with proteinase K at 37°C overnight. Proteinase K activity was inactivated by heating at 95°C for 10 minutes. For DNA extraction, standard methods were used: after incubation with proteinase K at 37°C overnight, the tissue was extracted twice in phenol and twice in chloroform, followed by ethanol precipitation.
Figure 1.

Microdissection of cholangiocarcinoma cells. The outlined areas (A) were microdissected (B) by the laser system (Palm MicrobeamSystem) and catapulted (C), as described in materials and methods.
Mutation analysis
All pre-PCR tissue was handled in an environment free of PCR products. All samples were coded and the investigator was blinded to all patient clinical details. Deparaffinised tissue was recovered by a 15 minute incubation with xylene followed by centrifugation for five minutes at 14 000 rpm twice. The tissue pellet was then washed twice in absolute ethanol followed by two washes in phosphate buffered saline. The pellet was incubated with 10 pellet volumes (approximately 500 μl) of lysis buffer (0.32 M sucrose, 10 mM Tris HCl, 1% (v/v) Triton X 100) and 0.2 volumes of proteinase K (final concentration 400 μg/ml) for 2–3 days at 37°C. DNA was phenol-chloroform extracted and precipitated in ethanol using conventional techniques. The resulting DNA pellet was resuspended in 50 μl TE buffer, pH 7.4 (10 mM Tris-HCl pH 7.4, 1 mM EDTA, pH 8.0). DNA samples were stored at −20°C.
For BRAF and KRAS mutation analysis, 25 HCC and 69 CC were used. In all cases, non-neoplastic liver tissue (obtained from tumour free resection margins and from distant areas of the liver) were analysed in parallel. All specimen were confirmed histologically.
Using these matched samples (tumour and normal liver from the same patient) screening for exon 2 to 18 BRAF mutations was performed with an ABI PRISM 3100 Genetic Analyser. PCR primers were designed to amplify the exon plus at least 50 bp of flanking intronic sequence according to previously published protocols.15 The primers were adopted from those published in the literature to omit analysing the BRAF pseudogene. The following primers were used:
exon 2: forward, GGAACACTGGCAGTTACTGTG; reverse, TTCCTAATCCCACCTCCTAAAA;
exon 3: forward, CAAAGAAACAGCAAAATGGTG; reverse, CAGGACAAAGTCCGGATTGA;
exon 4: forward, TTGCTCCCTTTACCTCTTATCAA; reverse, TTTCAATTCCCTAGGTTTTGG;
exon 5: forward, GCCCCTCGATAACCAATTTT; reverse, TCATCCATATTTCACATTCCCTA;
exon 6: forward, AACCCCCGGTTTTCATTTTA; reverse, CGTATGGAAGAAAAACCCTCA;
exon 7: forward, GAAGCTTCTGGGTTTTGCAC; reverse, AGTAGCATGTCGCCCAAGAG;
exon 8: forward, TCGTTACTCTGAATCTTATCTTCCA; reverse, TGAAAAATGGCACTTATTTCTGA;
exon 9: forward, TGGAAAATTCAGTGTTATCGCTAC; reverse, AAGGAAATAAGCAGCAAAGCA;
exon 10: forward, CCCAACCTTCTACCCCTGAT; reverse, GCAGTGCCGTAGAAATATGC;
exon 11: forward, TCCCTCTCAGGCATAAGGTAA; reverse, CGAACAGTGAATATTTCCTTTGAT;
exon 12: forward, TTGAAATGACACTTGGAGTAACAA; reverse, AGTTGCTACCACTGGGAACC;
exon 13: forward, TTGTAAGAATTGCTAAAGTTTGTCG; reverse, TCCAAAAGAATAGCAGCCAAA;
exon 14: forward, TTCGAGGCCAGAGTCCTTTA; reverse, GCTGTGGTATCCTGCTCTCC;
exon 15: forward, TCATAATGCTTGCTCTGATAGGA; reverse, GGCCAAAAATTTAATCAGTGGA;
exon 16: forward, GGTGTTTTAATGGTAAAAGCATTG; reverse, CGGTAAAATAAACACCAAGACG;
exon 17: forward, GGGTTTCCCACCATCTATGA; reverse, TGCTCAGAAATCTGTCTATGAATG;
exon 18: forward, CCACCCAGATTTTCATTCTTC; reverse, CCTTTTGTTGCTACTCTCCTGAA.
A total of 20 ng of genomic DNA from all test samples was amplified using standard PCR conditions and the resulting samples were then analysed on an ABI PRISM 3100 Genetic Analyser.18
The resulting traces were analysed to identify samples that produced a shift in peak migration relative to the matched normal control from the same individual or a standard normal control, indicating the presence of a putative sequence variation. Samples that produced a heteroduplex shift were directly sequenced on both strands using BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Perkin-Elmer/Cetus, Norwalk, Connecticut, USA) according to the manufacturer’s protocol, and analysed on an ABI PRISM 3100 Genetic Analyser.
The first exon of KRAS was amplified by PCR using primers designed to avoid amplification of the KRAS pseudogene. The primers used were 5′-ATTATAAGGCCTGCTGAAAATGAC TGA-3′ (upstream primer) and 5′-ATATGCATATTAAAACAAG ATTTACCT-CTA-3′ (downstream primer) giving a 155 bp product. Amplification was performed using a touchdown PCR technique15,16 from 63°C to 53°C over 10 cycles, followed by 30 cycles at 94°C, 53°C, and 72°C.
PCR products were purified using the Qiaquick PCR purification kit (Qiagen, Hilden, Germany) and sequenced using dye primer cycle sequencing and AmpliTaq polymerase FS on an Applied Biosystems DNA Sequencer (ABI PRISM 3100; Applied Biosystems-Perkin-Elmer/Cetus).
As a positive control for ras mutation analysis, DNA from colon carcinoma cell lines SW480 (Clontech, Palo Alto, California, USA) and HCT116 (American Type Culture Collection, ATCC, Rockville, Maryland, USA) with known KRAS mutations at codon 12 (GTT) and codon 13 (GAC), respectively, were used. Negative controls, without DNA, were run as controls for contamination.
If a mutation was detected, it was confirmed by amplification and sequencing of a fresh DNA sample using the upstream primer. Any sequences which proved difficult to read were re-amplified and re-sequenced.
Immunohistochemistry of active MAPK
Immunohistochemical analysis was performed as described previously.13 In all cases, tumour and non-neoplastic liver tissue was examined.
For immunohistochemical visualisation of active ERK, we used a polyclonal antibody (Anti-ACTIVE MAPK Ab; Promega, Madison, Wisconsin, USA) that specifically recognises the dually phosphorylated active form of MAPK (also known as p44/ERK1 and p42/ERK2) enzymes. The working dilution was 1:200.
Sections known to stain positively were included in each batch and negative controls were also performed by replacing the primary antibody with mouse or goat ascites fluid (Sigma-Aldrich Biochemicals, St Louis, Missouri, USA).
Statistics
Differences in frequencies between subgroups were analysed using the Kruskal-Wallis test and the Mann-Whitney U test for unpaired samples. Correlation coefficients were calculated according to Pearson, and χ2 statistics were used for contingency tables. Overall observed survival functions and probabilities were estimated using the Kaplan-Meier method. The log rank test was used to detect differences between survival curves for stratified variables. Identification of relevant prognostic factors was performed with univariate Cox regression analyses. The significance level was defined as p<0.05.
Median follow up of our patients was 510 days (range 50–2100). No patient was lost during follow up.
The medical records of all patients were re-examined to assess the status of disease at the closing date of the study (30 June 2002). At this time, 30 patients were still alive. All patients who died during the follow up period had intrahepatic and metastatic disease on their last visit to the oncological outpatients clinic. We concluded that death in these patients was related to HCC or CC.
RESULTS
BRAF gene alterations
Genomic DNA from HCC, CC, and corresponding liver tissue was analysed for BRAF gene mutations. In HCC, we failed to detect specific gene mutations. Excluding variants of unknown significance and germline polymorphisms found in liver tissue, somatic BRAF mutations were found in 15/69 CC (22%) (table 2 ▶). All mutations were within exons 11 and 15 (table 3 ▶), with a predominant nucleotide change (fig 2 ▶). A predominant mutation locus was in nucleotide 1796, which accounted for 11 of 15 mutations. This mutation leads to a substitution of valine by glutamic acid at position 599 (599 V→E). All mutations identified in CC were not present in the corresponding non-neoplastic liver tissue, indicating that the variants were somatically acquired mutations.
Table 2.
BRAF mutation in cholangiocarcinoma of the liver
| Patient No | Stage | Grade | Nucleotide | Amino acid substitution |
| 5 | I | 1 | 1796 T→A | 599 Valine→glutamate |
| 9 | II | 2 | 1403 G→T | 468 Glycine→alanine |
| 22 | II | 1 | 1796 T→A | 599 Valine→glutamate |
| 23 | IIIA | 3 | 1796 T→A | 599 Valine→glutamate |
| 30 | II | 2 | 1796 T→A | 599 Valine→glutamate |
| 32 | IIIA | 2 | 1786 C→G | 596 Leucine→valine |
| 33 | IIIC | 2 | 1796 T→A | 599 Valine→glutamate |
| 36 | IIIC | 2 | 1796 T→A | 599 Valine→glutamate |
| 37 | II | 2 | 1782 T→G | 594 Phenylalanine→leucine |
| 40 | II | 1 | 1796 T→A | 599 Valine→glutamate |
| 51 | I | 1 | 1403 G→A | 468 Glycine→glutamate |
| 53 | IIIA | 3 | 1796 T→A | 599 Valine→glutamate |
| 58 | II | 2 | 1796–97 TG→AT | 599 Valine→aspartate |
| 61 | II | 2 | 1796–97 TG→AT | 599 Valine→aspartate |
| 63 | II | 2 | 1796 T→A | 599 Valine→glutamate |
| 54 | Non-neoplastic liver | 1388 G→A | 463 Glycine→glutamate | |
Table 3.
KRAS mutation in cholangiocarcinoma
| Patient No | Stage* | Grade | Mutation | Amino acid substitution | p21ras (IHC†) |
| Codon 12 | GGT (wild-type) | Glycine | |||
| 1 | I | 1 | GGT→AGT | Serine | ++ |
| 2 | IIIA | 2 | GGT→GAT | Aspartate | ++ |
| 3 | II | 2 | GGT→GTT | Valine | ++ |
| 4 | IIIB | 3 | GGT→TGT | Cysteine | ++ |
| 7 | IIIA | 2 | GGT→GCT | Alanine | + |
| 8 | IIIC | 1 | GGT→TGT | Cysteine | ++ |
| 13 | IIIA | 1 | GGT→TGT | Cysteine | ++ |
| 14 | IIIB | 3 | GGT→GCT | Alanine | + |
| 17 | IIIA | 2 | GGT→GAT | Aspartate | ++ |
| 25 | IIIB | 2 | GGT→GCT | Alanine | + |
| 28 | IIIA | 2 | GGT→GTT | Valine | ++ |
| 29 | IIIC | 2 | GGT→TGT | Cysteine | ++ |
| 31 | IIIA | 2 | GGT→TGT | Cysteine | ++ |
| 33 | IIIC | 2 | GGT→GAT | Aspartate | ++ |
| 35 | IIIA | 1 | GGT→TGT | Cysteine | ++ |
| 38 | IIIA | 2 | GGT→AGT | Serine | +++ |
| 40 | II | 1 | GGT→GTT | Valine | ++ |
| 42 | II | 2 | GGT→TGT | Cysteine | ++ |
| 49 | I | 1 | GGT→TGT | Cysteine | ++ |
| 52 | IIIA | 3 | GGT→GAT | Aspartate | — |
| 59 | I | 1 | GGT→GTT | Valine | ++ |
| 61 | II | 2 | GGT→GCT | Alanine | ++ |
| 62 | IIIA | 3 | GGT→GCT | Alanine | +++ |
| 69 | IIIC | 3 | GGT→TGT | Cysteine | ++ |
| Codon 13 | |||||
| 6 | IIIA | 1 | GGC→GAC | Aspartate | +++ |
| 12 | I | 1 | GGC→TGC | Cysteine | ++ |
| 21 | II | 1 | GGC→CAT | Aspartate | + |
| 27 | IIIC | 2 | GGC→TGC | Cysteine | ++ |
| 34 | IIIA | 1 | GGC→GAC | Aspartate | +++ |
| 39 | II | 2 | GGC→CAT | Aspartate | + |
| 68 | IIIA | 2 | GGC→TGC | Cysteine | ++ |
| 5 | Non-neoplastic tissue | Codon 12 | GGT→GAT | Aspartate | + |
| 11 | Non-neoplastic tissue | Codon 13 | GGC→GAC | Aspartate | ++ |
*UICC 2002.17
†IHC, defined by immunohistochemistry.
Figure 2.

Mutation analysis of the BRAF gene. Electropherograms of the DNA sequences of patient Nos 9, 54, and 58 (same patients as in tables 2 and 3 ▶ ▶).
In one case (patient No 54), a mutation in nucleotide 1388 G→A was found in non-neoplastic tumour surrounding liver tissue, leading to a substitution of glycine to glutamate. This mutation was not present in the corresponding CC of the same patient. Mutation analysis of a lymph node from this patient, removed from the ligamentum hepatoduodenale, also revealed a wild-type status of BRAF. Identification of the nt1388 mutation in non-neoplastic liver prompted us to check codon 463 in all of our cases and also in removed lymph nodes. However, we detected the G→A nucleotide exchange only in the liver of patient No 54. Histopathological evaluation of the liver specimen of this patient revealed a strong cholangitis and a moderate steatosis with a slight pericellular fibrosis. The latter was assumed to be related to alcoholic consumption.
We failed to detect a significant correlation between the mutation status of BRAF, tumour stage or grade, or other histopathological factors (tumour size, vascular invasion, multiplicity, desmoplastic reaction).
KRAS status
PCR amplification and DNA sequencing enabled detection of heterozygous mutations in 31/69 CC (45%). Twenty four patients had a mutation of codon 12 and seven of codon 11. Ten of 31 mutations were G→A transitions. No patient had multiple mutations. In two cases, mutation of the KRAS gene was detected in non-neoplastic tumour surrounding liver tissue (table 3 ▶). One mutation was located at codon 12, the other at codon 13. The base pair changes in non-neoplastic tissue consisted of a G→A transition, producing an amino acid substitution of glycine for aspartic acid. These mutations were confirmed using different non-neoplastic liver tissue from patient Nos 5 and 11.
We failed to observe an association between KRAS mutation pattern and histopathological variables.
Of 15 tumours with BRAF mutations, 12 had a wild-type KRAS gene and three had mutated KRAS.
Immunohistochemistry
MAPK (p44/ERK1 and p42/ERK2) protein was detected immunohistochemically in all tumours to a variable extent. The number of positive cells within the tumour specimens varied from 12% to 89% (median 31%). Generally, immunoreactivity was confined to the tumour cell cytoplasm (fig 3A ▶). When the tumours were decoded for their BRAF status, all 19 CC with a BRAF mutation exhibited stronger immunostaining of the MAPK (p44/ERK1 and p42/ERK2) protein with a median of 69% positive tumour cells (fig 3B ▶). CC with a wild-type BRAF gene exhibited MAPK (p44/ERK1 and p42/ERK2) protein also. The number of positive cells was 33% (range 12–59) (fig 3C ▶).
Figure 3.
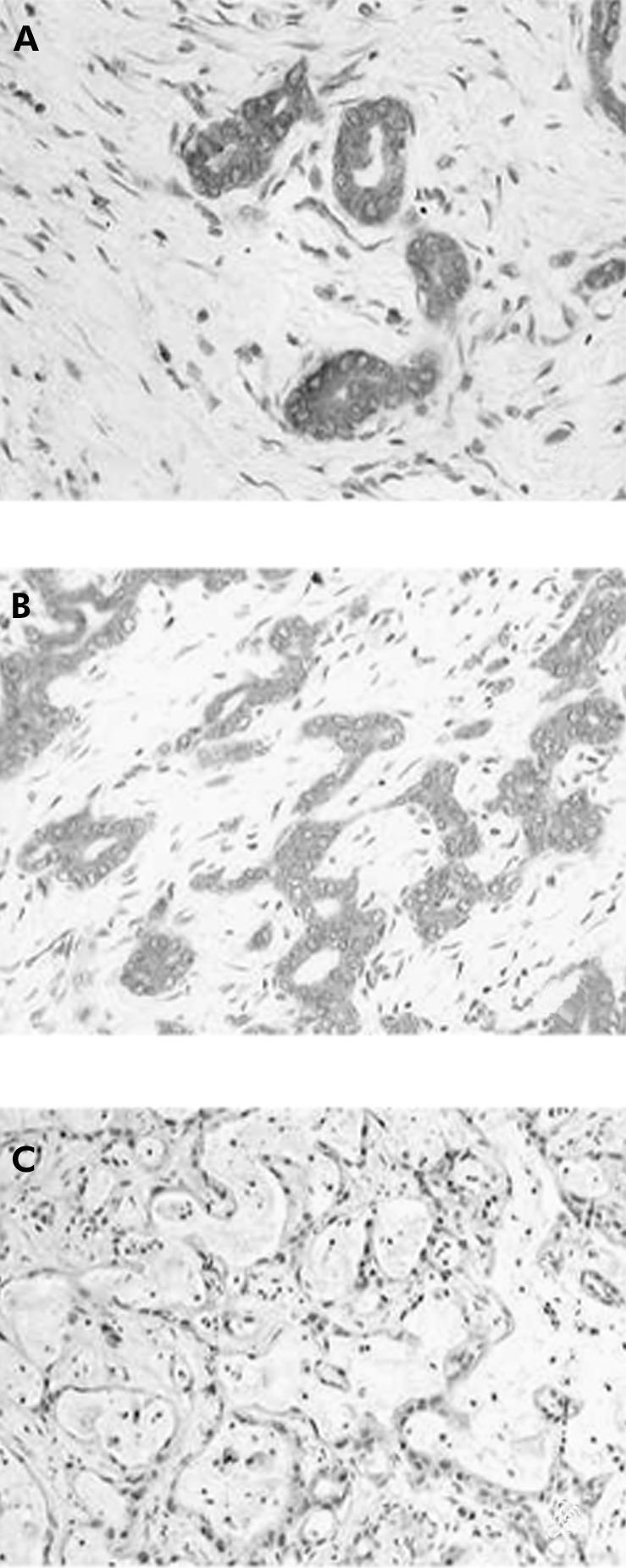
Immunostaining of the active MAPK protein. (A) Perinuclear staining (red reaction product) of tumour cells (original magnification ×60). (B) Tumour No 51 in table 2 ▶: well differentiated cholangiocarcinoma with a strong staining of active MAPK protein (original magnification ×20). (C) Tumour No 4 in table 3 ▶: poorly differentiated cholangiocarcinoma with partial mucinous differentiation. Tumour cells were positive for active MAPK to a lesser extent compared with (B) (original magnification ×20).
Survival rate
Survival analysis took into account the following variables: BRAF and KRAS status (mutated versus wild-type), UICC tumour stage (UICC 2002), grading, vascular invasion, multiplicity, desmoplastic reaction, necrosis, and patient age.
As expected, UICC stage, extent of the primary tumour (pT category), presence of lymph node metastases (pN category), and histological grade of tumour differentiation were significant prognostic parameters in univariate analysis. Neither BRAF nor KRAS were related to the prognosis of our patients. The odds ratios for all factors examined are given in table 1 ▶. In multivariate analysis, only extent of primary tumour (pT category) and lymph node status (pN category) had an independent prognostic impact.
DISCUSSION
In this study, we examined the frequency of mutations of BRAF and KRAS oncogenes in human HCC and CC. Both KRAS and BRAF are members of the RAS-RAF-MEK-ERK-MAP kinase pathway which mediates cellular response to growth signals.19 Whereas KRAS was examined in a variety of human malignancies,20 this is the first study of the mutational status of BRAF in liver tumours. In HCC, we failed to detect specific BRAF mutations. In contrast, CC showed specific BRAF mutations in approximately 21% and mutated KRAS in 44% of cases. Our data are in concordance with the literature, which described a trend for BRAF mutations in cancer types harbouring KRAS mutations.15 In HCC, KRAS mutations are rare events and therefore not a key event in heptocarcinogenesis.20 We also detected specific BRAF mutations in one case, and KRAS mutations in two cases of non-neoplastic liver tissue.
As was reported for melanomas, the highest frequency of BRAF mutations occurred at nucleotide 1796, leading to a T to A change at exon 15 of the BRAF gene.15 This mutation causes a single amino acid substitution (V599E and also V599D). Our mutation pattern observed in CC was nearly identical to those described by Davies et al in cell lines and primary human cancers.15 All mutations observed in CC were within exons 11 and 15 of the kinase domain, with a high prevalence of 1796 nucleotide mutations. These mutations are not only the predominant type in malignant melanoma but also in colon cancer and sarcoma.15 The affected amino acid residues are conserved in all three RAF genes through evolution and are identical at the equivalent positions in RAF1 and A-RAF. Transfection assays revealed that these mutations were active in vitro and stimulate the activity of the ERK pathway in vivo.15 Our data with a strong immunopositivity of the MAPK (p44/ERK1 and p42/ERK2) protein in CC with BRAF mutations may support these observations.
Furthermore, these kinase activated BRAF mutations possess the ability to induce transformation of NIH3T3 cells and exert tumorigeneity in nude mice experiments. Davies et al showed that RAS function was not required for the growth of cancer cell lines with the V599E mutation.15 According to our results, KRAS and BRAF mutations occur simultaneously in only three CC. In our series, the majority of CC exhibited either a BRAF or KRAS mutation; 23 CC had both wild-type BRAF and KRAS genes. As oncogenic KRAS activates wild-type BRAF, but mutated BRAF does not require KRAS for growth induction,6,9 a simultaneous BRAF and KRAS mutation in the same tumour may be redundant. Both KRAS and BRAF affect the same affector pathways (promoting cell survival by activating the MAPK pathway); a simultaneous “double mutation” may not confer a selection advantage for a single tumour cell.20 To prove this hypothesis, further studies are necessary, in particular to look for RAS and BRAF alterations in preneoplastic lesions or early tumour stages.
Interestingly, it was also shown by Davies and colleagues15 that the 1388 mutation, which was observed in non-neoplastic liver tissue of patient No 54 of our series, possesses kinase and transformation activity. Further studies are necessary, especially in livers of patients without CC, to evaluate the biological meaning of these findings.
The RAS-RAF-MEK-ERK-MAP kinase pathway can induce immortalisation, growth factor independent growth, insensitivity to growth inhibitory signals, ability to invade and metastasise, ability to secure nutrients by stimulating angiogenesis, avoidance of apoptosis, and altered response to chemotherapeutic drugs.4,5,10 Until now, very little evidence for tumour specific BRAF alterations has emerged. Apart from the report of Davies and colleagues,15 there are only episodic reports of BRAF overexpression, especially in colon cancer and lung cancer cell lines,7,10 but no genetic alterations have been found that could be linked to tumorigenesis. Many tumours exhibited alterations in the RAS-RAF-MEK-ERK-MAP kinase signalling pathways resulting in an increased signalling intensity via this cascade: more than 50% of colon tumours bear active mutants of KRAS and almost 70% of melanomas have a constitutively active B-RAF protein.19,20 In our series of CC, mutations of the kRAS oncogene occurred in 45%, which is in concordance with the literature.14 Together with 22% of BRAF mutations, the RAS-RAF-MEK-ERK-MAP kinase pathway showed a high frequency of alteration in CC. These data not only indicate that alterations of the RAS-RAF-MEK-ERK-MAP kinase pathway are important for the development of CC but may also highlight new strategies in the treatment of this disease.21 With an alteration rate of more than 60%, the RAS-RAF-MEK-ERK-MAP kinase cascade is by far the most common genetic alteration in CC. Therefore, inhibition of the RAS-RAF-MEK-ERK-MAP kinase pathway may be an important new therapeutic strategy in CC. The successful application of inhibitors of activated kinases was reported for STI571, an inhibitor of the BCR-ABL kinase, which is activated in chronic myeloid disorders or gastrointestinal stoma tumours.22
It has been reported that genetic alterations may influence the prognosis of cancer patients, especially for p53 and KRAS.16,23 In our series, neither the status of BRAF, KRAS, or both alterations influenced the survival of patients with CC. In our series, KRAS mutations were more common in advanced tumour stage. Due to strict selection criteria (only cases with primary curative (R0−) resection were examined, patients receiving liver transplantations were excluded), only a limited number of cases could be assessed for our study. Therefore, the actual prognostic value of KRAS or BRAF should be examined in a larger group of patients.
Acknowledgments
This paper was supported by the Bundesministerium für Bildung und Forschung (BMB+F), Interdisciplinary Centre for Clinical Research (IZKF) at the University of Leipzig (01KS9504/1, project D01).
Abbreviations
CC, cholangiocarcinoma
HCC, hepatocellular carcinoma
MAPK, mitogen activated protein kinase
ERK, extracellular signal regulated kinase
MEK, MAP/ERK
PCR, polymerase chain reaction
REFERENCES
- 1.Wittekind Ch. Hepatocellular carcinoma and cholangiocarcinoma. In: Hermanek P, Gospodarowicz MK, Henson DE, et al, eds. Prognostic Factors in Cancer. Berlin: Springer, 2002.
- 2.Taylor-Robinson SD, Toledano MB, Arora S, et al. Increase in mortality rates from intrahepatic cholangiocarcinoma in England and Wales 1968–1998. Gut 2001;48:816–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ding SF, Delhanty JD, Bowles L, et al. Loss of constitutional heterozygosity on chromosomes 5 and 17 in cholangiocarcinoma. Br J Cancer 1993;67:1007–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao YP, Studer EJ, Stravitz RT, et al. Activation of the Raf-1/MEK/ERK cascade by bile acids occurs via the epidermal growth factor receptor in primary rat hepatocytes. Hepatology 2002;35:307–14. [DOI] [PubMed] [Google Scholar]
- 5.van Deventer SJ. Small therapeutic molecules for the treatment of inflammatory bowel disease. Gut 2002; 50:(suppl 3):III47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao Y, Satoh T, Gao X, et al. RA-GEF-1, a guanine nucleotide exchange factor for Rap1, is activated by translocation induced by association with Rap1*GTP and enhances Rap1-dependent B-Raf activation. J Biol Chem 2001;276:28478–83. [DOI] [PubMed] [Google Scholar]
- 7.Mikula M, Schreiber M, Husak Z, et al. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1gene. EMBO J 2001;20:1952–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hakimi MA, Bochar DA, Chenoweth J, et al. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci U S A 2002;99:7420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia J, de Gunzburg J, Eychene A, et al. Thrombopoietin-mediated sustained activation of extracellular-regulated kinase in UT7-Mpl cells requires both Ras-Raf-1- and Rap1-B-Raf-dependent pathways. Mol Cell Biol 2001;21:2659–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peyssonnaux C, Eychene A. The Raf/MEK/ERK pathway: new concepts of activation. Biol Cell 2001;93:53–62. [DOI] [PubMed] [Google Scholar]
- 11.Kolch W. To be or not to be: a question of B-Raf? Trends Neurosci 2001;24:498–500. [DOI] [PubMed] [Google Scholar]
- 12.Hagemann C, Rapp UR. Isotype-specific functions of Raf kinases. Exp Cell Res 1999;253:34–46. [DOI] [PubMed] [Google Scholar]
- 13.Tannapfel A, Benicke M, Katalinic A, et al. Frequency of p16(INK4A) alterations and K-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut 2000;47:721–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiba T, Tsuda H, Pairokjul C, et al. Mutations of the p53 tumor suppressor gene and the ras gene family in intrahepatic cholangiocarcinomas in Japan and Thailand. Mol Carcinog 1993;8:312–18. [DOI] [PubMed] [Google Scholar]
- 15.Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–54. [DOI] [PubMed] [Google Scholar]
- 16.Hamilton SR, Aaltonen LA, eds. WHO: Pathology and Genetics. Tumours of the Digestive System. Lyon: IARC Press, 2000:173–80.
- 17.Sobin LH, Wittekind Ch, eds. UICC: TNM Classification of Malignant Tumors, 6th edn. New York: Wiley-Liss, 2002.
- 18.Tannapfel A, Wasner M, Krause K, et al. Expression of p73 and its relation to histopathology and prognosis in hepatocellular carcinoma. J Natl Cancer Inst 1999;91:1154–8. [DOI] [PubMed] [Google Scholar]
- 19.Kjoller L, Hall A. Signaling to Rho GTPases. Exp Cell Res 1999;253:166–79. [DOI] [PubMed] [Google Scholar]
- 20.Rajagopalan H, Bardelli A, Lengauer C, et al. RAF/RAS oncogenes and mismatch-repair status. Nature 2002;418:934. [DOI] [PubMed] [Google Scholar]
- 21.Kanno N, Lesage G, Phinizy JL, et al. Stimulation of alpha2-adrenergic receptor inhibits cholangiocarcinoma growth through modulation of Raf-1 and B-Raf activities. Hepatology 2002;35:1329–40. [DOI] [PubMed] [Google Scholar]
- 22.Hao D, Rowinsky EK. Inhibiting signal transduction: recent advances in the development of receptor tyrosine kinase and Ras inhibitors. Cancer Invest 2002;20:387–404. [DOI] [PubMed] [Google Scholar]
- 23.Tullo A, D’Erchia AM, Honda K, et al. New p53 mutations in hilar cholangiocarcinoma. Eur J Clin Invest 2000;30:798–803. [DOI] [PubMed] [Google Scholar]