

Abstract
Background: We present nine patients with progressive sclerosing cholangitis after septic shock.
Patients: All nine patients had previously required long term treatment in an intensive care unit for septic shock: two patients with polytrauma, five with burn injury, and two with extensive surgery. They were admitted to our hospital because of cholangitis. Endoscopic retrograde cholangiography revealed severe intrahepatic stenoses in all patients and liver biopsies showed typical signs of sclerosing cholangitis. No patient had pre-existing liver disease.
Results: Mean follow up time was 35 months. In patients with major bile duct stenoses (3/9), 12 endoscopic dilations were performed in total. In one patient, concrements were extracted and intermittent stenting was necessary. To date, 4/9 patients have rapidly developed liver cirrhosis. During follow up, 5/9 patients died: two after fulminant cholangitis, one after liver failure, one due to liver transplantation associated problems, and one after cerebral ischaemia. One patient has been registered for transplantation and the remaining three patients show no acute signs of liver failure.
Conclusions: Patients with sclerosing cholangitis, following septic shock, represent a new variant of vanishing bile duct disorders. In such patients liver disease rapidly progresses to cirrhosis. Endoscopic treatment may only transiently improve the course of the disease. Orthotopic liver transplantation is indicated in end stage disease.
Keywords: sclerosing cholangitis, sepsis, shock, endoscopic treatment, liver failure, biliary cirrhosis
A number of cholestatic syndromes, for example, primary biliary cirrhosis and primary sclerosing cholangitis (PSC), are characterised by progressive destruction of intrahepatic bile ducts. They are categorised as “vanishing bile duct” disorders (table 1 ▶).1 The presumed mechanisms causing injury and destruction of the bile ducts can be genetic, neoplastic, immunological, or ischaemic. Furthermore, toxic agents and infections can also cause biliary destruction. All of these cholestatic syndromes are characterised by the existence of cholangiocyte loss, associated with cholangiocyte proliferation, portal inflammation, and fibrosis.2 In general, they progress to cirrhosis and liver failure. In addition to the “vanishing bile duct” disorders, secondary sclerosing cholangitis (SSC) may be caused by bacterial cholangitis due to bile duct stones or biliary strictures after surgical treatment.3,4
Table 1.
Vanishing bile duct disorders
| Putative mechanisms | |
| Developmental-genetic | • Extrahepatic bile duct atresia |
| • Cystic fibrosis | |
| • Alpha1 antitrypsin deficiency | |
| • Progressive familiar intrahepatic cholestasis (types 1, 2, 3) | |
| Neoplastic | • Histiocytosis X |
| • Hodgkin’s disease | |
| Immunological | • Primary biliary cirrhosis |
| • Primary sclerosing cholangitis | |
| • Chronic graft versus host disease | |
| • Sarcoidosis | |
| • Hepatic allograft rejection | |
| • Autoimmune cholangitis | |
| Infectious | • Ascending cholangitis (bacterial) |
| • Cytomgegalovirus | |
| • Hepatitis C virus | |
| • Cryptosporidia | |
| • Retrovirus type 3 | |
| Toxic | • Paraquat |
| • Chlorpromazine | |
| • Floxuridine after intra-arterial infusion | |
| • Formaldehyde/20% hypertonic saline, injected into hyatid cyst | |
| • Amoxicillin | |
| Ischaemic | • Intra-arterial infusions or embolisations |
| • Hepatic allograft rejection | |
| Unclassified | • Idiopathic adulthood ductopenia |
Here we present nine patients who developed secondary sclerosing cholangitis after septic shock associated with long term ventilation. In addition to reporting this new variant, the endoscopic and medical treatment as well as the clinical course and outcome of these patients are discussed.
PATIENTS AND METHODS
During the period August 1995 to February 2002, nine patients with severe SSC were seen in our hospital, Department of Gastroenterology, University of Heidelberg. All nine patients had previously required long term intensive care treatment for septic shock. All required long term ventilation (15–60 days) and catecholamines. Two developed septic shock after polytrauma caused by road accidents (one with splenic rupture followed by splenectomy, multiple rib fractures, and contusion of the lung and liver, and the other with subarachnoid bleeding, multiple rib fractures, haemothorax, vertebral body fractures, and open lower leg fracture). Two patients developed septicaemia after extensive surgery (one after an aortocoronary bypass and the other after sigmoidectomy for colonic carcinoma followed by a prolonged abdominal wound dehiscence). The remaining five patients suffered severe burn injury (second to third degree, 23–45% of body surface). They were treated in a burn unit over a long period due to septic shock, and received multiple necrotomies and split skin grafts. All patients developed elevated liver enzymes and cholestasis 7–67 days after the trauma (table 2 ▶). Later they were admitted to our hospital with signs of cholangitis and upper abdominal pain.
Table 2.
Patient data
| Patient No 1 | Patient No 2 | Patient No 3 | Patient No 4 | Patient No 5 | Patient No 6 | Patient No 7 | Patient No 8 | Patient No 9 | |
| Sex | Female | Female | Male | Male | Female | Female | Male | Male | Male |
| Age at presentation (y) | 69 | 59 | 16 | 60 | 59 | 47 | 55 | 73 | 68 |
| Cause of admission | Polytrauma | Polytrauma | Burn injury II-III (44% body surface) |
Burn injury II-III (33% body surface) |
Burn injury II-III (34% body surface) |
Burn injury II-III (45% body surface) |
Burn injury II-III (24% body surface) |
Sigma carcinoma, sigmoidectomy | Aorto-coronary bypass |
| Ventilation (days) | 44 | 32 | 15 | 40 | 60 | 60 | 59 | 18 | 42 |
| 1st documentation of cholestasis* (days after injury/surgery) | 7 | 7 | 19 | 19 | 52 | 67 | 63 | 24 | 25 |
| Time until 1st admission† (months) | 0.4 (11 days) | 0.6 (19 days) | 15.5 | 1.7 | 31 | 17.8 | 28 | 1.3 | 3.9 |
| Bilirubin (mg/dl)‡ | 17.2 | 7.6 | 2 | 31 | 1.5 | 8 | 3.2 | 14.3 | 7.4 |
| AP (U/l)‡ | 1064 | 579 | 1637 | 2989 | 419 | 1905 | 1094 | 3451 | 1798 |
| GGT (U/l)‡ | 236 | 420 | 428 | 858 | 45 | 359 | 430 | 782 | 448 |
*Elevated bilirubin, alkaline phosphatase (AP), and gamma glutamyl transpeptidase (GGT).
†Interval between first documentation of cholestasis and first visit to the Department of Gastroenterology.
‡Laboratory at first visit to the Departement of Gastroenterology.
The diagnosis of sclerosing cholangitis was based on endoscopic retrograde cholangiopancreaticography (ERCP). No patient had pre-existing liver disease. Viral and autoimmune hepatitis, haemochromatosis, Wilson’s disease, and alcohol abuse were excluded. All eight patients, who were tested for autoimmune parameters, had negative antinuclear antibodies, antimitochondrial antibodies, smooth muscle autoantibodies, antibodies against soluble liver antigen, antibodies against liver-kidney microsomal antigen, antineutrophil cytoplasmic antibodies, and hypergammaglobulinaemia. As assessed by colour assisted duplex sonography, the hepatic artery showed patency in all cases and thrombosis or obstruction of the hepatic veins and the portal vein were ruled out. No patient had signs of diarrhoea, colonic bleeding, or colitis at any stage.
Prior to admission at our hospital, cholecystectomy (CHE) was performed on three patients: two patients (patient Nos 5 and 6) had cholecystolithiasis and underwent CHE (five months and 17 months after the injury). One patient (No 1) had suspected cholecystitis (without cholecystolithiasis) and was operated 28 days after the injury. All operations were aimed at improving the hepatobiliary disease and cholestasis but all were unsuccessful.
RESULTS
Patient clinical data on admission to our hospital are shown in table 2 ▶. All patients had hyperbilirubinaemia ranging from 1.5 to 31.0 mg/dl (mean 10.2 (8.9) mg/dl). Elevated gamma glutamyl transpeptidase (GGT) and alkaline phosphatase (AP) levels were the predominant laboratory findings: mean AP 1660 l (986) U/l, mean GGT 445 (235) U/l. At initial presentation to our hospital, three patients had already developed liver cirrhosis (patients No 5, 6, and 7).
ERCP showed severe intrahepatic stenoses and rarefaction of the small bile ducts in all patients. In addition, 3/9 patients (patient Nos 3, 6, and 7) had one or more dominant stenoses of the right or left hepatic duct. In patient Nos 3 and 6, the right hepatic lobe was already atrophic. Only one patient (patient No 7) showed strictures and irregularities of the common bile duct, and concrements were found. Pancreatic ducts were entirely normal in all nine patients.
Needle biopsies of the liver were available from seven patients. One patient died before biopsy could be conducted but was autopsied. One patient refused a liver biopsy. All biopsies showed ductular bile duct proliferation (fig 1 ▶), a minimal portal lymphocytic inflammatory infiltrate, and portal fibrosis with some periductular fibrosis (fig 2 ▶). In 3/7 biopsies (patient Nos 5, 6 and 7), micronodular cirrhosis was found.
Figure 1.

Liver biopsy of patient No 9. Immunostaining for cytokeratin 7. Ductules express cytokeratin 7. Some hepatocytes express bile duct cytokeratin 7, showing bile duct proliferations arising from ductular metaplasia of periportal hepatocytes.
Figure 2.

Liver biopsy of patient No 9. Staining with Masson-Goldner showed periportal fibrosis and a chronic inflammatory infiltrate with some neutrophils.
Follow up and treatment
Mean follow up time was 35 months (range 1.5–74). Treatment and clinical outcome are summarised in table 3 ▶. All patients received ursodeoxycholic acid (UDCA) (8.9–15.6 mg/kg body weight) and antibiotics. In all three patients with dominant bile duct stenoses (patients No 3, 6, and 7) endoscopic sphincterotomy of the papilla (EPT) was performed, followed by balloon dilation of the stenosis. Altogether, 12 endoscopic dilations and in one patient (patient No 7) intermittent stenting were necessary. In patient No 7, common duct stones were extracted after EPT and extracorporeal shock wave lithotripsies. In patient Nos 1 and 9, large amounts of sludge were removed after EPT. Biochemical improvement of cholestasis was achieved in eight of our nine patients (table 4 ▶). In patient No 4, liver enzymes and jaundice did not improve. In addition to the patients who presented with liver cirrhosis on admission to our hospital (patient Nos 5, 6, and 7), one further patient (patient No 3) developed cirrhosis after admission.
Table 3.
Clinical data
| Patient No 1 | Patient No 2 | Patient No 3 | Patient No 4 | Patient No 5 | Patient No 6 | Patient No 7 | Patient No 8 | Patient No 9 | |
| Biopsy | − | − | + | + | + | + | + | + | + |
| Cholecystolithiasis | − | − | − | − | + | + | − | − | − |
| Cholecystectomy | − | − | − | − | + | + | − | − | − |
| Common duct stones | − | − | − | − | − | − | + | − | − |
| EPT/dilatations | +/− | − | +/1 | − | − | +/5 | +/6 | − | +/− |
| Liver cirrhosis* | − | − | 57 | − | 12 | 17 | 18 | − | − |
| Liver transplantation (months after injury/surgery) | − | − | (listed) | − | − | − | 67 | − | − |
| Death (months after injury/surgery) | Sepsis, multiorgan failure (1.5) | − | − | Sepsis, multiorgan failure (4) | Hepatic coma (55) | − | Right heart failure (74) | − | Cerebral ischaemia (10) |
| Autopsy | + | − | − | − | − | − | + | − | − |
| Follow up (months/yrs after injury) | 1.5/0.1 | 22/1.8 | 74/6.2 | 4/0.3 | 55/4.6 | 73/6.1 | 74/6.2 | 2.6/0.2 | 10/0.8 |
EPT, endoscopic sphincterotomy of the papilla.
*First diagnosis months after injury/surgery.
Table 4.
Mean laboratory parameters before and after treatment
| AP (U/l) | Bilirubin (mg/dl) | |
| Dilatation group* | ||
| Before treatment | 1806 | 11.1 |
| After treatment | 1117 | 2.3 |
| Group with EPT and sludge extraction† | ||
| Before treatment | 637 | 15 |
| After treatment | 102 | 6.5 |
| UDCA only group‡ | ||
| Before treatment | 2017 (1675)§ | 17.7 (14.2)§ |
| After treatment | 1236 (1114 )§ | 13.7 (7.3)§ |
*Patients with endoscopic dilatations and ursodeoxycholic acid (UDCA) treatment (Nos 3, 6, and 7).
†Patients with endoscopic sphincterotomy of the papilla (EPT), sludge extraction, and UDCA treatment (Nos 1 and 9).
‡Patients with UDCA treatment only (Nos 2, 4, 5, and 8).
§Data calculated without patient No 4 who did not respond to therapy.
AP, alkaline phosphatase.
During follow up, 5/9 patients died. Two patients (patient Nos 1 and 4) showed a fulminant course of cholangitis and died of sepsis and multiorgan failure (fig 3 ▶). One of four patients with cirrhosis (patient No 5) died of hepatic coma and liver failure (fig 4 ▶). Patient No 7 was transplanted 66 months after the trauma and died due to transplantation associated problems (fig 5 ▶). Patient No 9 died after multiple cerebral ischaemias and severe bronchopneumonia. To date, 4/9 patients are still alive (patient Nos 2, 3, 6, and 8). Patient No 3 has been registered in our transplant programme due to deteriorating liver function and several episodes of oesophageal variceal bleeding. At present, the remaining three patients (patient Nos 2, 6, and 8) are still in a relatively stable condition without acute signs of liver failure.
Figure 3.
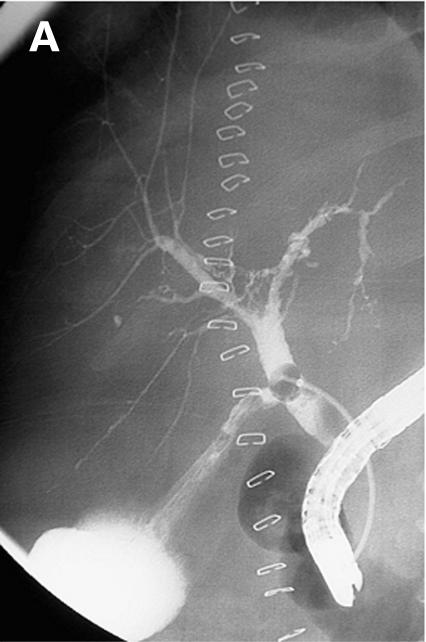

Patient No 1. (A) Endoscopic retrograde cholangiopancreaticography (ERCP) showing multiple strictures in the intrahepatic biliary system. (B) Clinical course with laboratory data. EPT, endoscopic sphincterotomy, CHE, cholecystectomy, AP, alkaline phosphatase.
Figure 4.
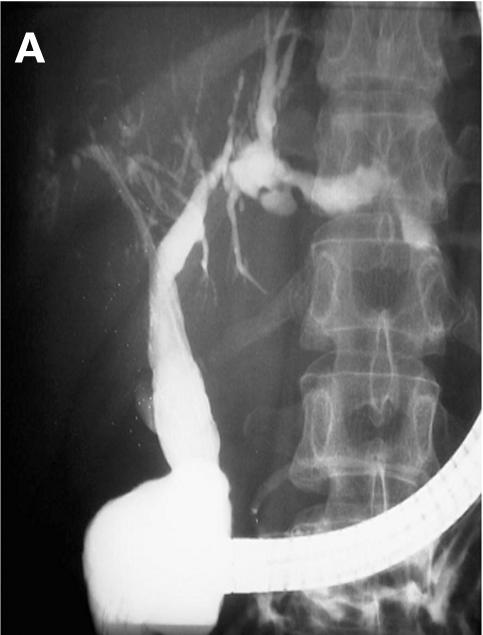

Patient No 5. (A) Endoscopic retrograde cholangiopancreaticography (ERCP) showing multiple intrahepatic strictures in the biliary system. (B) Clinical course with laboratory data. AP, alkaline phosphatase.
Figure 5.
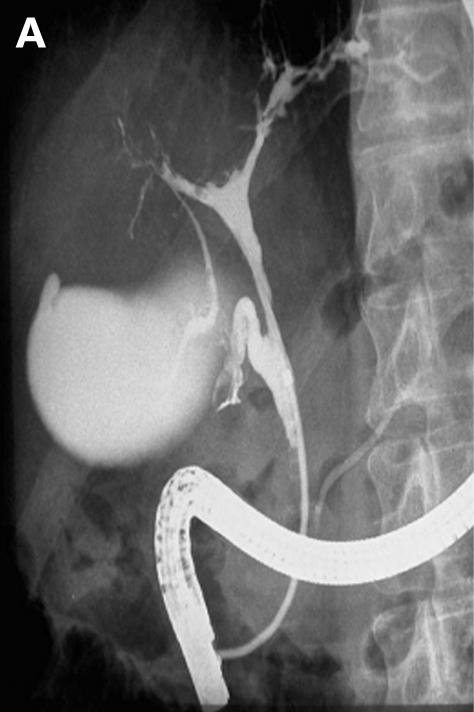

Patient No 7. (A) Endoscopic retrograde cholangiopancreaticography (ERCP) showing intrahepatic strictures and dominant stenoses in the right biliary system, the common bile duct shows irregularities of diameter. (B) Clinical course with laboratory data. L, lithotripsy, D, dilatation, E, endoprosthesis, OLT, orthotopic liver transplantation; AP, alkaline phosphatase.
DISCUSSION
We have reported nine cases of rapidly progressive SSC after septic shock due to burn injury, polytrauma, or extensive surgery. To date, only a preliminary report of patient No 5, describing SSC after burn injury, has been published.5 Recently, Scheppach et al described three patients with sclerosing cholangitis after bacterial extrahepatic infection that rapidly progressed to biliary cirrhosis.6 In our patients, however, it seems very unlikely that bacterial cholangitis was the primary cause of the progressive bile duct destruction as endoscopic and antibiotic treatment did not lead to sustained improvement. In general, patients with bacterial cholangitis have biliary obstruction, and endoscopic and antibiotic treatment lead to rapid improvement of the disease. This was not the case in our nine patients.
In all of our patients, the diagnosis of sclerosing cholangitis was based on ERCP which showed multiple ductual stricturesand stenoses, and typical liver biopsy findings with bile duct proliferation, portal and periductular fibrosis, and a minimal portal lymphocytic inflammatory infiltrate. The clinical course of our patients varied: two patients (patients No 1 and 4) developed fulminant cholangitis with sepsis, rapidly leading to multiple organ failure and death. The others presented with slower progression and with recurrent episodes of cholangitis. In patients with dominant bile duct stenoses, endoscopic interventions were performed, and cholestasis and clinical symptoms were improved intermittently (Nos 3, 6, and 7). In one third of our patients (Nos 5, 6, and 7), cirrhosis developed very rapidly (1–1.5 years), and in one patient (No 3) cirrhosis developed approximately five years after the injury. Two patients (Nos 2 and 6) had markedly elevated serum bilirubin levels but without clinical signs of liver failure or cirrhosis to date. The observation period for patient No 8 has been too short to decide whether or not cirrhosis will develop.
Secondary sclerosing cholangitis is a progressive disease leading to secondary biliary cirrhosis. In such patients, orthotopic liver transplantation (OLT) is the only therapeutic option.3,4,6 Several mechanisms have been identified that perpetuate SSC, including infectious cholangitis, human immunodeficiency virus infection, and cytomegalovirus infection. SSC may also occur due to ischaemic related causes—for example, after arterial embolisation or hepatic arterial thrombosis, as well as a result of stenosis of the hepatic artery after OLT.3,7–9 In patient Nos 1 and 2, who developed bile duct strictures and cholestasis very rapidly after polytrauma, temporary ischaemia due to liver contusion cannot be excluded. The exact mechanisms responsible for the development of SSC or ductopenia in our patients however remain unclear.
Firstly, all of our patients required long term intensive care treatment because of the development of shock and sepsis, and they received total parenteral nutrition (TPN) over extended periods. During and after long term TPN, cholestasis or steatosis are frequently observed but these disorders are mostly mild and reversible.10–13 A causal relation to the formation of severe bile duct stenoses is unlikely. Furthermore, the incidence of bile stone formation is significantly associated with TPN when administered for more than three weeks.10,12,14–17 Concrements in the common bile duct were found in one patient (No 7). Patient Nos 5 and 6 had cholecystolithiasis without evidence of gall stones in the bile duct. A causal role for gall stones may be discussed in these three patients but can be excluded in all of the other patients. It seems more likely that gall stones formed secondary to bile duct stenosis.
Bile duct injury may be related to sepsis and shock. Sepsis causes endothelial injury to the gastrointestinal tract with ensuing gut barrier failure. Several factors may play a role. These include direct toxic effects on microvascular endothelium caused by bacterial endotoxins and lipopolysaccharides. Furthermore, a lipopolysaccharide induced massive release of proinflammatory cytokines can cause destruction of the mucosal wall.18,19 Septicaemia or shock are both known to cause intestinal ischaemia that results in gut endothelial injury. Neutrophils, adherent to the endothelial surface, cause increased microvascular permeability and local oedema, especially during the reperfusion phase after splanchnic hypoperfusion.18, 20,21 Gut barrier failure follows and may induce translocation of bacteria and endotoxins from the intestinal lumen into the portal blood, and possibly into the bile ducts.18,22,23 Following cholangitis, postinflammatory biliary strictures may develop, especially if bile flow is impaired due to TPN. Moreover, thermal injuries in particular can induce selective vasoconstriction of hepatic arterial blood flow. This additional hepatic ischaemia and critical reduction of hepatic oxygen delivery may lead to sclerosing cholangitis.24 These mechanisms may all contribute to the development of SSC but it is still unclear why it progressed so quickly in most of our patients.
The biliary alterations in SSC reported here, together with their clinical significance, may be compared with complications seen in PSC. Long term results of several trials in patients with PSC suggest an improvement in clinical symptoms and liver function tests after regular endoscopic treatment.25–27 Recently, it has been shown that UDCA treatment, in combination with repeated endoscopic interventions and opening of bile duct stenoses, may further improve survival and the clinical outcome of patients with PSC.28,29 The same effect might be achieved in our patients with SSC. The combination of endoscopic and medical treatment with UDCA caused a transient biochemical improvement in cholestasis in eight of our nine patients. Our data indicate that once secondary biliary cirrhosis has occurred, endoscopic treatment may temporarily improve liver function. However, despite this, progression of the disease was still noted in all of our patients.
In summary, patients with SSC after septic shock represent a new entity of vanishing bile duct disorders. The clinical course varies considerably, but in most cases liver disease rapidly progresses to cirrhosis. Our department represents a referral institution for biliary diseases but it is impossible to tell how many patients with septic shock develop such a biliary disorder. Endoscopic treatment in combination with UDCA may transiently improve the clinical course but if end stage liver disease develops orthotopic liver transplantation is indicated.
Abbreviations
PSC, primary sclerosing cholangitis
SSC, secondary sclerosing cholangitis
ERCP, endoscopic retrograde cholangiopancreaticography
CHE, cholecystectomy
GGT, gamma glutamyl transpeptidase
AP, alkaline phosphatase
UDCA, ursodeoxycholic acid
EPT, endoscopic sphincterotomy of the papilla
OLT, orthotopic liver transplantation
TPN, total parenteral nutrition
REFERENCES
- 1.Desmet VJ. Vanishing bile duct disorders. In: Boyer JL, Ockner RK, eds. Progress in Liver Diseases, vol X. Philadelphia: WB Saunders; 1992:89–121. [PubMed]
- 2.Desmet VJ, van Eyken P, Roskams T. Histopathology of vanishing bile duct diseases. Adv Clin Path 1998;2:87–99. [PubMed] [Google Scholar]
- 3.Amor A, Chapoutot C, Michel J, et al. Secondary sclerosing cholangitis. Presse Med 1995;24:948–52. [PubMed] [Google Scholar]
- 4.Loinaz C, Gonzalez EM, Jimenez C, et al. Long-term biliary complications after liver surgery leading to liver transplantation. World J Surg 2001;25:1260–3. [DOI] [PubMed] [Google Scholar]
- 5.Schmitt M, Kölbel CB, Müller MK, et al. Sklerosierende Cholangitis nach Verbrennung. Z Gastroenterol 1997;35:929–34. [PubMed] [Google Scholar]
- 6.Scheppach W, Druge G, Wittenberg G, et al. Sclerosing cholangitis and liver cirrhosis after extrabiliary infections: report on three cases. Crit Care Med 2001;29:438–41. [DOI] [PubMed] [Google Scholar]
- 7.Sebagh M, Farges O, Kalil A, et al. Sclerosing cholangitis following human orthotopic liver transplantation. Am J Surg Pathol 1995;19:81–90. [DOI] [PubMed] [Google Scholar]
- 8.Tsimoyiannis EC, Grantzis E, Moutesidou K, et al. Secondary sclerosing cholangitis: after injection of formaldehyde into a hyatid cyst in the liver. Eur J Surg 1995;161:299–300. [PubMed] [Google Scholar]
- 9.Heneghan MA, Sylvestre PB. Cholestatic diseases of liver transplantation. Semin Gastrointest Dis 2001;12:133–47. [PubMed] [Google Scholar]
- 10.Quigley E, Marsh M, Shaffer J, et al. Hepatobiliary complications of total parenteral nutrition. Gastroenterology 1993;104:286–301. [DOI] [PubMed] [Google Scholar]
- 11.Müller M. Hepatische Komplikationen bei parenteraler Ernährung. Z Gastroenterol 1996;34:36–40. [PubMed] [Google Scholar]
- 12.Baker F, Rosenberg W. Hepatic complications of total parenteral nutrition. Am J Med 1987;82:489–97. [DOI] [PubMed] [Google Scholar]
- 13.Machnik G. Histologische Veränderungen des Lebergewebes bei Cholestase. Z Gastroenterol 1993;31(suppl 2):7–10. [PubMed] [Google Scholar]
- 14.Holzbach R. Gallbladderstasis: consequence of long-term parenteral hyperalimentation and risk factor for cholelithiasis. Gastroenterology 1983;84:1055–8. [PubMed] [Google Scholar]
- 15.Messing B, Bories C, Kunstlinger F, et al. Does total parenteral nutrition induce gallbladder sludge formation and lithiasis? Gastroenterology 1983;84:1012–19. [PubMed] [Google Scholar]
- 16.DeBoer SY, Masclee AAM, Jebbing MCW. Effect of intravenous fat on cholecystokinin secretion and gallbladder motility in man. JPEN J Parenter Enteral Nutr 1992;16:16–19. [DOI] [PubMed] [Google Scholar]
- 17.Pitt H, King I, Mann L. Increased risk of cholelithiasis with prolonged total parenteral nutrition. Am J Surg 1983;145:106–11. [DOI] [PubMed] [Google Scholar]
- 18.Brophy C. Gastrointestinal vascular ischemic syndromes. Curr Opin Gen Surg 1993;225–31. [PubMed]
- 19.McCarthy Pastores S, Katz D, Kvetan V. Splanchnic ischemia and gut mucosal injury in sepsis and the multiple organ dysfunction syndrome. Am J Gastroenterol 1996;91:1697–710. [PubMed] [Google Scholar]
- 20.Cicalese L, Caraceni P, Nalesnik M, et al. Oxygen free radical content and neutrophil infiltration are important determinants in mucosal injury after rat small bowel transplantation. Transplantation 1996;62:161–6. [DOI] [PubMed] [Google Scholar]
- 21.Moore F, Moore E. Evolving concepts in the pathogenesis of postinjury multiple organ failure. Surg Clin North Am 1995;75:257–77. [DOI] [PubMed] [Google Scholar]
- 22.Deitch EA. The role of intestinal barrier failure and bacterial translocation in the development of systemic infection and multiple organ failure. Arch Surg 1990;125:403–8. [DOI] [PubMed] [Google Scholar]
- 23.Baron P, Traber LD, Traber DL, et al. Gut failure and translocation following burn and sepsis. J Surg Res 1994;57:197–204. [DOI] [PubMed] [Google Scholar]
- 24.Tadros T, Traber DL, Herndon DN. Hepatic blood flow and oxygen consumption after burn and sepsis. J Trauma 2000;49:101–8. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, Schutz S, England R, et al. Endoscopic therapy of sclerosing cholangitis. Hepatology 1995;21:661–7. [PubMed] [Google Scholar]
- 26.Wagner S, Gebel M, Trautwein C, et al. Endoscopic management of biliary tract strictures in primary sclerosing cholangitis. Endoscopy 1996;28:546–51. [DOI] [PubMed] [Google Scholar]
- 27.Baluyut AR, Sherman S, Lehman GA, et al. Impact of endoscopic therapy on the survival of patients with primary sclerosing cholangitis. Gastrointest Endosc 2001;53:308–12. [DOI] [PubMed] [Google Scholar]
- 28.Stiehl A, Rudolph G, Sauer P, et al. Efficacy of ursodeoxycholic acid treatment and endoscopic dilation of major duct stenoses in primary sclerosing cholangitis. J Hepatol 1997;26:560–6. [DOI] [PubMed] [Google Scholar]
- 29.Stiehl A, Rudolph G, Klöters-Plachky P, et al. Development of dominant bile duct stenoses in patients with primary sclerosing cholangitis treated with ursodeoxycholic acid: outcome of endoscopic treatment. J Hepatol 2002;36:151–6. [DOI] [PubMed] [Google Scholar]