

Abstract
Background: Hepatic adenosine triphosphate (ATP) levels are an accurate reflection of functioning hepatic mass following surgical resections and acute liver injury.
Objective: To determine whether hepatic ATP levels can serve as a non-invasive means of documenting progression of chronic liver disease to cirrhosis.
Methods: In vivo phosphorus-31 magnetic resonance spectroscopy (31P MRS) was performed in three animal models of chronic liver disease. Sixty six adult Sprague- Dawley rats were subjected to either thioacetamide, carbon tetrachloride (CCl4), or common bile duct ligation (CBDL) to induce liver disease (n=35, 21, and 10, respectively). Serial MRS examinations, blood samples, and liver biopsies (when appropriate) were obtained throughout and/or on completion of the study.
Results: Over the course of the chronic liver disease, a progressive decrease in hepatic ATP levels was consistently observed in each model. The findings were most striking when end stage liver disease (cirrhosis) was established. The reduction in hepatic ATP levels correlated with significant changes in serum albumin concentrations (CCl4 and CBDL models) and the extent of hepatocyte loss seen histologically (all models).
Conclusion: The results of this study indicate that during progression of chronic liver disease to cirrhosis, there is a progressive reduction in hepatic ATP levels. In addition, changes in hepatic ATP levels correlate with changes in liver function and histology. Thus hepatic 31P MRS provides a non-invasive means of documenting the severity and progression of parenchymal and cholestatic models of chronic liver disease in rats.
Keywords: liver, magnetic resonance spectroscopy, liver disease, cirrhosis, rat
The majority of chronic liver disorders have the potential to progress in an insidious manner to cirrhosis.1,2 Cirrhosis is a diffuse condition, characterised by loss of functioning hepatic mass, extensive fibrosis, and reconstruction of hepatic lobules into abnormal nodular patterns.3 Initially, the condition is asymptomatic and associated with normal liver function tests as the residual liver is able to compensate for hepatocyte necrosis and changes in hepatic architecture. Eventually, the disease and structural distortions become sufficiently extensive to result in hepatic decompensation and death from liver failure or complications thereof.1
Rates of progression of chronic liver diseases to cirrhosis are highly variable. For some patients, cirrhosis can develop within 1–2 years while for others, 2–3 decades may be required.4 This variability highlights the importance of being able to accurately document the rate of disease progression, which often impacts on decisions regarding therapeutic interventions. To date, liver biopsies remain the gold standard whereby assessments of liver disease are conducted.5–7 However, a number of problems surround this procedure including: (1) it is invasive, occasionally painful, and often feared by patients, (2) complications can be life threatening, (3) it is prone to sampling error, and (4) the histological interpretation is subjective.8–10 Alternative means of documenting the extent of chronic liver disease and dysfunction include traditional blood tests (serum aminotransferases, bilirubin, and albumin levels, and plasma prothrombin times) and classification systems such as Child-Pugh scores.11 Quantitative liver function tests can also be employed; these include various measurements of hepatic blood flow, clearance rates, and hepatic metabolism of exogenous agents.12 Unfortunately, the applicability of these tests is limited by inaccuracies, cumbersome data collection, and/or difficulties in interpretation.11,13
Phosphorus-31 magnetic resonance spectroscopy (31P MRS) is a radiological technique that provides direct biochemical information about the tissue under consideration. This technique permits non-invasive simultaneous detection and quantitation of several cytosolic phosphorus containing compounds involved in energy metabolism (adenosine triphosphate (ATP) and inorganic phosphate (Pi)) and membrane phospholipid metabolism (phosphomonoesters (PME) and phosphodiesters (PDE)).
Previous studies in our laboratory have demonstrated that hepatic ATP levels in particular accurately reflect the extent of hepatic disease and dysfunction following graded surgical resections and in animal models of acute liver disease.14,15 Reports by other groups have suggested that changes in PME resonance may also be indicative of the extent of disease.16–18 The aim of the present study was to determine whether levels of hepatic metabolites (ATP, Pi, PME, and/or PDE), as detected by quantitative31P MRS, reflect the severity and progression of chronic liver disease to cirrhosis in rats.
MATERIAL AND METHODS
Experimental animals
Adult male Sprague-Dawley rats were maintained on Purina rat chow and water ad libitum until initiation of the study. All animals were kept in identical housing units on a 12 hour light- dark cycle. The number of rats assigned to each experimental group was based on published data regarding the consistency of the model employed, anticipated death rates, and number of analyses outlined in the experimental protocol. This study was approved by the University of Manitoba animal ethics committee.
Thioacetamide induced liver cirrhosis
Thioacetamide (TAA) cirrhosis was induced in 35 rats (150–250 g) by administering TAA (Sigma Chemicals, St Louis, Missouri, USA; 30 mg/100 ml) in their drinking water over a period of six months.19,20 Serial MRS examinations were performed at baseline and at two, four, and six months during TAA exposure. Following MRS examinations at each time, a subset of rats (4–12 per group) was sacrificed, and blood and liver samples collected. For animals that underwent serial examinations (n=10) over the six month period, percutaneous liver biopsies were performed following each MRS examination.
Twelve isocaloric fed rats not exposed to TAA served as controls. Serial MRS examinations were also performed on this group. However, rats were only sacrificed at baseline and six months.
Carbon tetrachloride induced liver cirrhosis
This group consisted of 27 rats weighing 100–120 g. All animals had free access to water containing 0.35 g/l phenobarbital (Abbott Laboratories, Canada) starting two weeks prior to and continuing throughout carbon tetrachloride (CCl4) administration.21 CCl4 (Sigma Chemicals) was given intragastrically twice a week to 21 rats for 8–9 weeks. CCl4 was diluted in 0.5 ml of corn oil and doses were continuously adjusted on the basis of individual body weight, as described by Dupin and colleagues.22 Six control rats underwent the same procedure, but received only corn oil intragastrically.
MRS examinations were performed on all animals 10–14 days after their last CCl4 or corn oil treatment. Following each MRS examination, animals were sacrificed, and blood and liver samples were collected.
Common bile duct ligation induced cirrhosis
Common bile duct ligation (CBDL) was used to produce secondary biliary cirrhosis in 10 rats (240–270 g).23 Briefly, under ether anaesthesia, a 4 cm incision was made just below the xiphoid process. The bile duct was isolated, a double ligation of the common bile duct was performed, and the bile duct was severed between the ligatures. The abdomen was subsequently closed and the animal allowed to recover.
Serial MRS examinations and blood sample collections were performed on each rat at baseline (preoperatively), and on days 12, 27, and 46 post surgery. Following measurements on day 46, animals were sacrificed and liver samples were collected. MRS examinations, and blood and liver sample collections, were also performed on day 46 on an additional group of age matched rats (n=4) not subjected to surgery.
31P magnetic resonance spectroscopy
All MRS examinations were performed on an MSLX Bruker Biospec spectrometer equipped with a 7 Tesla 21 cm bore horizontal magnet (Bruker, Karlsruhe, Germany). Prior to MR examinations, animals were fasted overnight. All MR examinations were conducted between 8:00 am and 12:00 noon where animals were anaesthetised with isoflurane and placed on their right side on a 31P/1H doubly tunable double ring surface coil (15/40 mm diameter) operating at 121.5/300 MHz. A small vial containing methylene diphosphonic acid (MDP) was placed near the centre of the coil to assist with subject positioning during MR imaging, calibration of the RF field strength at the region of interest (ROI), and for quantitation of metabolite concentrations. Snapshot-Flash MR images were acquired in the axial plane with repetition time (TR)/echo time (TE)=3.7/2.2 ms, slice thickness of 2 mm, field of view (FOV) 8 cm×8 cm, and a matrix size of 128×128. Localised shimming on the liver was performed using a VOSY sequence with a 15×15×25 mm3 (lateral, vertical, and axial dimensions, respectively) voxel using TE=15 ms and a mixing time of 20 ms. The frequency of the coil was then tuned to phosphorus and the 90° pulse length was determined for the MDP reference vial near the centre of the coil. A non-localised fully relaxed spectrum of the MDP reference was acquired for measurements of coil loading. Based on the characteristics of the B1 field of the surface coil, a 90° pulse length was determined at the centre of the ROI for subsequent localised spectroscopy. Localised 31P liver spectra were acquired using two dimensional chemical shift imaging with a chemical shift selective suppression of the residual MDP resonance. Data acquisition parameters were FOV= 8.85 cm (horizontal) × 6.0 cm (vertical), 14 averages, TR=3 s, matrix size=8×8 zero filled to 16×16, acquisition size=1 K, zero filled to 2 K, and sweep width=4840 Hz. Data processing was accomplished using WIN-MRI and WIN-NMR software (Bruker-Franzen Analytik GmbH, Bremen, Germany). Briefly, the free induction decay underwent 30 Hz exponential line broadening prior to Fourier transformation, and the resulting spectra were processed with manual phase and baseline correction. Peaks were registered relative to α-ATP resonance (−10 ppm) which served as an internal chemical shift reference. Finally, peak integrals were calculated by Gaussian curve fitting with all signals treated as singlets. Corrections for minor contributions of metabolite signal arising from overlying skeletal muscle were performed. Based on the percentage of phosphocreatine contamination in the liver spectra, relative amounts of muscle signal contributing to each metabolite were calculated according to previous published data.24,25 These values were then subtracted from the appropriate integral to give a “pure” liver reading.
For quantitiation of hepatic metabolites, phantom experiments were performed as described by Meyerhoff and colleagues.26 A 250 ml flask containing 50 mM sodium phosphate served as a phantom, on which identical MRS examinations were performed regularly throughout the experiment. The various metabolite concentrations were determined by the equation27:
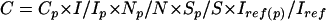 |
where C=absolute metabolite concentration in mmol/l; Cp=concentration of phantom solution used for calibration in mmol/l; I, Ip=corresponding signal integrals; N, Np=corresponding number of signal averages; S, Sp=corresponding saturation factors calculated from measured T1 times; and Iref, Iref(p)=corresponding signal integral of reference sample
Literature T1 values for liver were used in the calculation for saturation factors.28
Hepatic ATP levels were determined using the peak area of the β-ATP resonance, which is free of contributions from other phosphorylated adenosine species.29
Liver function
Sera were isolated from collected blood samples, and serum aspartate aminotransferase (AST) and albumin levels were measured using commercial kits (Sigma).
Histology and quantitative morphological analysis of liver tissue
Fixed tissue specimens were blocked in paraffin, cut, and stained with haematoxylin-eosin and van Gieson (collagen) stains. Slides were staged (0–IV) by a pathologist (blinded to the study groups) for hepatic fibrosis according to the following scale: stage 0, no fibrosis; stage I (mild fibrosis), fibrous expansion around portal tracts or central veins; stage II (moderate fibrosis), septa extending into the liver lobule; stage III (moderate fibrosis), bridging fibrosis (portal-portal, central-central, or portal-central linkage); and stage IV (cirrhosis), parenchymal nodules surrounded by fibrous septa and disturbed hepatic architecture.
Images of van Gieson stained sections were captured with a Spot Cooled Color Digital Camera and Spot Software v2.2 (Diagnostic Instruments, Inc., Sterling Heights, Michigan, USA). A 40× power microscopic field of the liver tissue was displayed on a colour monitor as an 800×600 pixel image. Proportional areas of liver tissue and fibrosis were determined using a computerised image analysis system (Image Pro Plus Software Package; Silver Spring, Maryland, USA) and the liver cell area/whole tissue area ratio was then calculated. The procedure was carried out over 10–15 FOV on each tissue section excluding large vessels and large portal tracts. The mean value from all fields was expressed as a percentage defined as the “liver cell area ratio” (LCAR), which represents liver cell volume per unit of liver tissue.
Statistical evaluation
Results are expressed as mean (SEM). Analysis of variance with Tukey-Kramer correction was used to examine differences between groups. Spearman correlations were performed to test for associations between non-parametric test variables, with a p value of less than 0.05 considered significant.
RESULTS
TAA model
Animals in the TAA series tended to gain weight over the course of the disease, as rats at stages 2, 3, and 4 weighed more than those at stages 0 and 1 (table 1 ▶). Liver weights were also greater at stages 3 and 4 than at the preceding stages.
Table 1.
Rat body and liver weights at various stages of chronic liver disease
| Group | n | Body weight (g) | Liver weight (g) |
| Thioacetamide series | |||
| Stage 0 | 9 | 341 (15) | 14.8 (0.9) |
| Stage 1 | 4 | 270 (9.0) | 11.1 (0.6) |
| Stage 2 | 4 | 410 (15)* | 15.0 (1.6) |
| Stage 3 | 7 | 423 (15)* | 19.1 (1.5)† |
| Stage 4 | 9 | 415 (15)* | 19.7 (1.6)† |
| Carbon tetrachloride series | |||
| Stage 1 | 6 | 433 (12) | 11 (0.5) |
| Stage 4 | 10 | 413 (11) | 13 (1.0) |
| Common bile duct ligation series | |||
| Baseline | 8 | 283 (7.0) | — |
| Cholestasis | |||
| 11 days | 9 | 295 (7.0) | — |
| 26 days | 9 | 344 (10)‡ | — |
| 46 days | 7 | 376 (24)§ | 20.0 (2.1) |
| Age matched controls | 4 | 461 (12) | 14.1 (0.9) |
Values are means (SEM).
*p<0.05 versus stages 0 and 1; †p<0.05 versus stage 1; ‡ p<0.05 versus baseline; § p<0.05 versus baseline and age matched controls.
Table 2 ▶ provides the results of serum AST and albumin determinations at various stages of disease. Similar to the reports of others, levels of serum AST and albumin did not differ with stage of disease.30,31 However, LCAR decreased with progressive disease reaching significantly lower levels at stages 2, 3, and 4.
Table 2.
Serum AST and albumin levels, and LCAR in control rats and rats at various stages of thioacetamide induced chronic liver disease
| Stage | Serum AST (U/l) | Serum albumin (g/l) | LCAR (%) |
| 0 | 65.7 (14.4) | 38.5 (0.9) | 98.5 (0.2) |
| 1 | 72.4 (24.0) | 42.6 (2.6) | 98.2 (0.1) |
| 2 | 59.2 (10.0) | 40.6 (1.8) | 93.6 (2.0)* |
| 3 | 76.5 (15.0) | 40.6 (1.0) | 90.1 (2.5)† |
| 4 | 89.1 (14.4) | 42.0 (0.8) | 86.4 (2.5)‡ |
AST, aspartate aminotransferase; LCAR, liver cell area ratio.
*p<0.05 versus stages 0 and 4; †p<0.05 versus stages 0 and 1; ‡p<0.05 versus stages 0, 1, and 2.
Representative sections of liver tissue and corresponding localised 31P MR spectra from rats with normal livers (stage 0) and those with stages 2, 3, and 4 of TAA induced chronic liver disease are provided in fig 1 ▶. In addition to the fibrosis and cirrhosis traditionally described for late stages of chronic liver disease, regions of ductular cholangiocellular proliferation were frequently present in TAA treated rats with stages 3 and 4 disease.
Figure 1.

Representative histological section of the liver and corresponding hepatic 31P magnetic resonance spectrum from (A) a healthy rat (stage 0), and rats with thioacetamide induced chronic liver disease (B) stage 2, (C) stage 3, and (D) stage 4. Van Gieson, ×20 (see methods for description of histological staging). PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate. PCr, phosphocreatine resonance arising from abdominal wall muscle.
Hepatic 31P MR spectra from rats with normal livers (stage 0), and stages 1 and 2 disease were similar. Once disease progressed to stages 3 and 4, a significant decrease in the signal of phosphorylated metabolites was seen in the 31P MR spectra. Concentrations of the various phosphorylated metabolites are presented in table 3 ▶. Hepatic ATP levels decreased with progression of disease such that significantly lower levels were documented once rats progressed to stages 3 and 4. Levels of Pi fluctuated with progression of disease but significantly lower levels were detected at stages 2, 3, and 4 compared with levels at stages 0 and 1. PME levels displayed a downward trend with disease progression while PDE levels remained unchanged.
Table 3.
Concentrations (mM) of hepatic phosphorylated metabolites in control rats and rats at various stages of thioacetamide induced chronic liver disease
| Stage | PME | Pi | PDE | ATP |
| 0 | 3.68 (0.17) | 1.55 (0.07) | 1.05 (0.09) | 3.04 (0.12) |
| 1 | 3.63 (0.40) | 1.90 (0.28) | 1.13 (0.15) | 2.82 (0.04) |
| 2 | 3.05 (0.17) | 0.91 (0.07)* | 1.23 (0.11) | 2.61 (0.14) |
| 3 | 3.03 (0.88) | 1.21 (0.08)* | 1.28 (0.12) | 2.21 (0.17)† |
| 4 | 2.55 (0.17) | 0.94 (0.06)* | 1.11 (0.09) | 2.18 (0.15)† |
Values are means (SEM). PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate.
*p<0.05 versus stages 0 and 1; †p<0.05 versus stage 0.
Correlation analysis (table 4 ▶) revealed no significant associations between these hepatic phosphorylated metabolite levels and serum AST but Pi levels did correlate with serum albumin concentrations (r=−0.48, p<0.004). Significant correlations also existed between the LCAR index and hepatic ATP (r=0.56, p<0.001) and PME (r=0.46, p<0.01) levels.
Table 4.
Correlations between hepatic phosphorylated metabolites, serum AST and albumin levels, and LCAR in thioacetamide induced chronic liver disease
| Correlation (r) | p Value | |
| Serum AST | ||
| PME | 0.01 | 0.97 |
| Pi | −0.02 | 0.92 |
| PDE | −0.10 | 0.59 |
| ATP | 0.01 | 0.95 |
| Serum albumin | ||
| PME | −0.21 | 0.24 |
| Pi | −0.48 | 0.004 |
| PDE | 0.09 | 0.65 |
| ATP | −0.25 | 0.15 |
| LCAR | ||
| PME | 0.46 | 0.01 |
| Pi | 0.29 | 0.14 |
| PDE | −0.12 | 0.52 |
| ATP | 0.56 | 0.001 |
AST, aspartate aminotransferase; LCAR, liver cell area ratio; PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate.
CCl4 model
Body and liver weights among rats exposed to CCl4 were similar to those of controls (table 1 ▶) as were serum AST concentrations (table 5 ▶). However, serum albumin concentrations in rats with stage 4 disease were significantly lower than in rats with stage 1 disease (30.2 (0.6) v 35.2 (1.2) g/l, respectively; p<0.05). Stage 4 livers also had a lower LCAR index than those at stage 1 (83.9 (1.2)% v 98.6 (0.2)%, respectively; p<0.05).
Table 5.
Serum AST and albumin levels, and LCAR in control rats and rats with carbon tetrachloride induced chronic liver disease
| Stage | Serum AST (U/l) |
Serum albumin (g/l) | LCAR (%) |
| 1 | 60.98 (10.70) | 35.23 (0.63) | 98.56 (0.21) |
| 4 | 75.14 (13.07) | 30.23 (1.20)* | 83.86 (1.22)* |
AST, aspartate aminotransferase; LCAR, liver cell area ratio.
*p<0.05 versus stage 1.
Animals treated for 8–9 weeks with CCl4 had complete cirrhosis (stage 4) while those treated with corn oil alone displayed non-specific histological changes. Presented in fig 2 ▶ are representative histological sections and hepatic31P MR spectra from cirrhotic and control rats. Table 6 ▶ shows concentrations of hepatic phosphorylated metabolites in CCl4 treated rats and controls. Similar to TAA experiments, animals with stage 4 disease had decreased hepatic ATP levels compared with rats with stage 1 disease. Hepatic Pi and PME levels demonstrated a trend towards lower levels with increasing severity of disease but this did not reach statistical significance. PDE levels remained unchanged.
Figure 2.

Representative histological section of liver and corresponding hepatic 31P magnetic resonance spectrum from (A) a control rat and (B) a rat with carbon tetrachloride induced cirrhosis. Van Gieson, ×20 (see methods for description of histological staging). PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate. PCr, phospho- creatine resonance arising from abdominal wall muscle.
Table 6.
Concentrations (mM) of hepatic phosphorylated metabolites in control rats and rats with carbon tetrachloride induced chronic liver disease
| Stage | PME | Pi | PDE | ATP |
| 1 | 3.61 (0.39) | 0.94 (0.11) | 0.90 (0.16) | 3.33 (0.20) |
| 4 | 3.35 (0.18) | 1.09 (0.06) | 0.81 (0.08) | 2.71 (0.10)* |
Values are means (SEM).
PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate.
*p<0.05 versus stage 1.
Correlations between hepatic phosphorylated metabolites, serum AST, and albumin concentrations and LCAR in CCl4 treated rats are presented in table 7 ▶. Hepatic ATP levels correlated with both serum albumin (r=0.53, p<0.02) and LCAR (r=0.63, p<0.006).
Table 7.
Correlations between hepatic phosphorylated metabolites, serum AST and albumin levels, and LCAR in rats with carbon tetrachloride induced chronic liver disease
| Correlation (r) | p Value | |
| Serum AST | ||
| PME | 0.04 | 0.86 |
| Pi | 0.13 | 0.62 |
| PDE | 0.01 | 0.95 |
| ATP | 0.26 | 0.30 |
| Serum albumin | ||
| PME | 0.00 | 0.99 |
| Pi | −0.34 | 0.17 |
| PDE | 0.19 | 0.35 |
| ATP | 0.53 | 0.02 |
| LCAR | ||
| PME | 0.25 | 0.32 |
| Pi | −0.24 | 0.35 |
| PDE | 0.29 | 0.17 |
| ATP | 0.63 | 0.006 |
AST, aspartate aminotransferase; LCAR, liver cell area ratio; PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate.
CBDL model
As shown in table 1 ▶, animals in the CBDL series followed a similar trend to those described for the TAA group in that rats with more advanced disease (days 26 and 46) weighed more than those at baseline or with early disease (day 12). Age matched controls weighed more than their day 46 cholestatic counterparts. Liver weights were similar between the two groups (table 1 ▶). Significant elevations in serum AST and reductions in serum albumin concentrations and the LCAR index occurred in cholestatic rats over the course of the disease (table 8 ▶).
Table 8.
Serum AST and albumin levels, and LCAR in control rats and rats with chronic cholestatic liver disease following common bile duct ligation
| Time | Serum AST (U/l) | Serum albumin (g/l) | LCAR (%) |
| Baseline | 73.9 (18.4) | 48.2 (1.4) | — |
| 12 days | 237 (29)* | 42.0 (2.3) | — |
| 27 days | 217 (38)* | 33.5 (2.8)‡ | — |
| 46 days | 325 (98)† | 27.5 (1.9)† | 70.7 (5.8)* |
| Aged matched controls | 33.0 (5.4) | 39.4 (1.1)§ | 96.0 (0.7) |
AST, aspartate aminotransferase; LCAR, liver cell area ratio.
*p<0.05 versus aged matched controls; †p<0.05 versus baseline, 12 days, and aged matched controls; ‡p<0.05 versus baseline and 12 days; §p<0.05 versus baseline and 46 days
Representative sections of liver tissue and the corresponding liver31P MR spectra from a control rat and a rat with chronic cholestasis are displayed in fig 3 ▶. At 46 days post bile duct ligation, hepatic lesions which included marked hepatocyte necrosis, inflammation, bile duct proliferation, and fibrosis, were in keeping with biliary cirrhosis. A summary of the levels of hepatic phosphorylated metabolites documented during the course of the chronic cholestatic liver disease is presented in table 9 ▶. Hepatic ATP levels progressively decreased, with significantly lower levels occurring at 47 days post surgery (fig 3 ▶). Correlation analysis (table 10 ▶) revealed that hepatic ATP levels correlated with serum AST (r=−0.57, p<0.002), albumin (r=0.42, p<0.01) concentrations, and LCAR (r=0.71, p<0.02).
Figure 3.

Representative histological section of liver and corresponding hepatic 31P magnetic resonance spectrum from (A) a control rat and (B) a rat with cholestatic (common bile duct ligation) induced cirrhosis. Van Gieson, ×40 (see methods for description of histological staging). PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate. PCr, phosphocreatine resonance arising from abdominal wall muscle.
Table 9.
Concentrations (mM) of hepatic phosphorylated metabolites in control rats and rats with chronic cholestatic liver disease following common bile duct ligation
| Time | PME | Pi | PDE | ATP |
| Baseline | 3.05 (0.24) | 1.64 (0.11) | 0.94 (0.15) | 3.13 (0.22) |
| 12 days | 3.21 (0.19) | 1.56 (0.10) | 1.01 (0.07) | 2.69 (0.22) |
| 27 days | 3.20 (0.28) | 1.72 (0.16) | 0.98 (0.09) | 2.60 (0.30) |
| 46 days | 2.64 (0.30) | 1.43 (0.17) | 0.97 (0.19) | 1.66 (0.13)* |
| Age matched controls | 3.04 (0.13) | 1.30 (0.03) | 1.49 (0.37) | 3.28 (0.34) |
Values are means (SEM).
PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate.
*p<0.05 versus baseline, aged matched controls, 12 day, and 27 day cholestatic rats.
Table 10.
Correlations between hepatic phosphorylated metabolites, serum AST and albumin levels, and LCAR in rats with common bile duct ligated induced cholestatic liver disease
| Correlation (r) | p Value | |
| Serum AST | ||
| PME | −0.08 | 0.70 |
| Pi | 0.14 | 0.50 |
| PDE | −0.36 | 0.11 |
| ATP | −0.57 | 0.002 |
| Serum albumin | ||
| PME | −0.23 | 0.19 |
| Pi | −0.02 | 0.91 |
| PDE | 0.13 | 0.52 |
| ATP | 0.42 | 0.01 |
| LCAR | ||
| PME | 0.24 | 0.51 |
| Pi | 0.39 | 0.26 |
| PDE | 0.53 | 0.14 |
| ATP | 0.71 | 0.02 |
AST, aspartate aminotransferase; LCAR, liver cell area ratio; PME, phosphomonoesters; Pi, inorganic phosphate; PDE, phosphodiesters; ATP, adenosine triphosphate.
DISCUSSION
Chronic liver disease can result from various causes which operate through different pathophysiological pathways and elicit distinct patterns of hepatic injury.1,2,32 However, the general course and outcome of chronic hepatic injury, as described by progressive fibrosis and eventually cirrhosis, remains constant.1,6 In a similar manner, the present study reports consistent alterations in hepatic metabolism throughout the course of hepatocellular and cholestatic chronic liver diseases to cirrhosis. Animals subjected to TAA, CCl4, and CBDL induced chronic liver disease all displayed progressive reductions in hepatic ATP levels with disease progression.
Although the mechanism(s) responsible for the decline in hepatic ATP levels remains to be determined, gradual loss of viable hepatocytes is likely to be an important contributing factor. As the total amount of these cells per unit volume of liver decreases, MR detectable signal from that volume will also decrease. Indeed, image analysis of tissue sections taken at various stages of disease revealed a progressive decrease in the LCAR index that correlated with decreases in hepatic ATP levels. Thus during early phases of disease, when hepatocyte loss is offset by active hepatic regeneration, only minor changes in hepatic ATP were detected.33,34 Once more advanced disease was established, and when regenerative activity becomes impaired,35 hepatic ATP levels decreased and the decrease was proportional to the loss of functional hepatic tissue.
Another possible explanation relates to altered hepatic bioenergetics, namely increased energy expenditure as liver disease progresses.36 With the reduction in total volume of viable liver tissue, residual hepatocytes must expend more energy to maintain normal hepatic function and engage in compensatory liver regeneration. Eventually, the remnant hepatocyte population is incapable of meeting the increasing demands and both energy depletion and hepatic insufficiency ensue. Supporting this explanation were the decreases in serum albumin concentrations and hepatic ATP levels in CCl4 treated and CBDL rats. That serum albumin concentrations were maintained during TAA induced chronic liver disease is in keeping with the findings of other investigators who reported no or few changes in liver function tests in TAA treated animals.30,31,37 Further studies are required to examine the compensatory mechanism in this model.
Disturbed hepatic bioenergetics has also been ascribed to the capillarisation of hepatic sinusoids during the development of cirrhosis.38 In normal livers, the sieve-like structure of the sinusoidal endothelium and the freely accessible perisinusoidal space allow for rapid and high exchange of substrates and nutrients between the vascular compartment and hepatocytes. During fibrogenesis and the development of cirrhosis, a barrier of connective tissue and extracellular matrix is deposited in the perisinusoidal space.39,40 By limiting hepatocyte access to nutrients, high energy substrates, and oxygen, this barrier may impair hepatic energy metabolism.38,41
The magnitude of hepatic ATP levels at end stage disease varied considerably between the models of chronic liver disease, being highest in CCl4 treated rats (ATP 2.71 mM), lowest in CBDL rats (ATP 1.66 mM), and intermediate in TAA treated rats (ATP 2.18 mM), despite similar levels of LCAR in each model. This disparity may be explained by the different histological features present. Specifically, CCl4 administration produced the typical pattern of cirrhosis with bridging fibrotic septa, disorganised parenchymal architecture, regenerative nodules, and mild periportal bile ductal proliferation. Although the same features were present in the TAA model, TAA exposed rat livers also contained multiple cholangiofibromas. In the CBDL model, a different pattern of cirrhosis was present where extensive bile ductal proliferation extended beyond periportal regions and replaced much of the normal parenchyma. Given that the LCAR index includes areas covered by both parenchymal and non-parenchymal cells and that luminal space occupies much of the area enclosed by bile ducts, the disparity observed in the hepatic ATP levels could reflect differences in viable hepatocyte populations. It is important to note that the differences between the various models should not affect the results of serial examinations within the same model or individual subjects within the group.
An alternative explanation for differences in absolute ATP levels relates to the heterogeneity of the liver lobule and patterns of zonal injury induced by the various models. Periportal hepatocytes within zone 1 of the liver lobule have sufficiently high oxygen tension that they are capable of generating energy by both glycolytic and oxidative metabolism, whereas the less aerobic perivenous hepatocytes predominantly utilise glycolytic pathways of energy generation.42,43 Having access to both aerobic and anaerobic pathways, and thereby a selection of fatty acid and carbohydrate substrates, periportal hepatocyte are capable of generating more energy on a per mole basis than those in the perivenous region.44,45 Hence one would predict that lesions specific to zone 1 of the liver lobule would create a greater bioenergetic disturbance than lesions localised in zone 3. The results from the present study support this hypothesis as CBDL injury predominantly involves zone 1 while CCl4 involves zone 3 and TAA is a more diffuse injury, involving zone 1–3 hepatocytes.
Inorganic phosphate, another marker of tissue bioenergetics, displayed a trend towards lower levels with more advanced stages of chronic liver disease. However, significant decreases were only detected among TAA treated rats. As with hepatic ATP, lower levels of hepatic Pi likely result from reduced hepatocyte mass. However, unlike ATP, these changes are attenuated by certain metabolic activities within the functioning remnant liver. Specifically, increased energy expenditure perpetuates the hydrolysis of high energy phosphate bonds which in turn liberates inorganic phosphate species. Accumulation of Pi due to enhanced metabolic activity and reduced recycling back to purine/pyrimidine moieties would in turn contribute to the Pi signal. Indeed, increases in Pi have been observed during high energy activities such as liver regeneration following partial hepatectomy.15,46
Information regarding phospholipid membrane metabolism may also be obtained from the PME and PDE resonances in the 31P MR spectrum. Both resonances are multicomponent peaks containing contributions from several metabolites.47 The PME resonance consists of membrane phospholipid precursors, phosphoenthaolamine (PE), and phosphocholine (PC), and several phosphorylated glycolytic (G) intermediates. Conversely, PDE resonance is made up of metabolites involved in phospholipid membrane degradation, GPC, and GPE, as well as signal arising from the phospholipid bilayers of the endoplasmic reticulum.47–49 An increase in PME levels accompanied by a decrease in PDE levels has traditionally been interpreted to reflect increased phospholipid membrane turnover due to enhanced cell proliferation.50 Previous studies in human patients with cirrhosis have reported increased ratios of hepatic PME/ATP and PME/PDE.16,51 Additional high resolution MRS experiments performed on tissue extracts have demonstrated that indeed significant elevations in hepatic PE and PC and reductions in GPE and GPC occur within the cirrhotic liver.18 These findings are interpreted to reflect enhanced cell turnover as the cirrhotic liver attempts to regenerate.17,18
Contrary to previous reports, in this study PME levels did not increase nor did PDE levels decrease in any of the models employed. These findings raise the possibility that hepatic phospholipid membrane activity may differ in rat models of cirrhosis versus cirrhosis in humans. An alternative explanation relates to differences in the field strength at which the present study was performed compared with previous studies as PDE resonance is known to contain a magnetic field dependent component.48,49
In conclusion, the results of the present study indicate that hepatic ATP levels correlate with biochemical evidence of hepatic dysfunction and histological evidence of loss of functioning hepatocytes and progressive disease. The results also support the concept that regardless of the aetiology, progressive liver disease has a significant effect on hepatic bioenergetic integrity. Given these observations, hepatic 31P MRS holds promise as a non-invasive means of documenting the extent and progression of liver disease.
Acknowledgments
This work was supported by grants from the Health Sciences Centre Foundation and the Canadian Liver Foundation. Financial assistance from the Multimedia Advanced Computational Infrastructure (MACI) project is appreciated. Special thank you to Dr Sahar Al-Haddad from the Department of Pathology for staging histological sections.
Abbreviations
31P-MRS, phosphorus-31 magnetic resonance spectroscopy
ATP, adenosine triphosphate
Pi, inorganic phosphate
PME, phosphomonoesters
PDE, phosphodiesters
TAA, thioacetamide
CCl4, carbon tetrachloride
CBDL, common bile duct ligation
MDP, methylene diphosphonic acid
ROI, region of interest
TR, repetition time
TE, echo time
FOV, field of view
AST, aspartate aminotransferase
LCAR, liver cell area ratio
PE, phosphoenthaolamine
PC, phosphocholine
REFERENCES
- 1.Chung RT, Jaffe DL, Friedman LS. Complications of chronic liver disease. Crit Care Clin 1995;11:431–63. [PubMed] [Google Scholar]
- 2.Williams EJ, Iredale JP. Liver cirrhosis. Postgrad Med J 1998;74:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Popper H. Pathologic aspects of cirrhosis. A review. Am J Pathol 1977;87:228–64. [PMC free article] [PubMed] [Google Scholar]
- 4.Hoofnagle JH. Hepatitis C: the clinical spectrum of disease. Hepatology 1997;26(suppl 1):15–20S. [DOI] [PubMed] [Google Scholar]
- 5.Degos F, Benhamou JP. Liver Biopsy. Oxford: Oxford University Press, 1999.
- 6.Desmet VJ, Gerber M, Hoofnagle JH, et al. Classification of chronic hepatitis: diagnosis, grading and staging. Hepatology 1994;19:1513–20. [PubMed] [Google Scholar]
- 7.Perrillo RP. The role of liver biopsy in hepatitis C. Hepatology 1997;26(suppl 1):57–61S. [DOI] [PubMed] [Google Scholar]
- 8.Bravo AA, Sheth SG, Chopra S. Liver biopsy. N Engl J Med 2001;344:495–500. [DOI] [PubMed] [Google Scholar]
- 9.Nord HJ. Biopsy diagnosis of cirrhosis: blind percutaneous versus guided direct vision techniques—a review. Gastrointest Endosc 1982;28:102–4. [DOI] [PubMed] [Google Scholar]
- 10.Piccinino F, Sagnelli E, Pasquale G, et al. Complications following percutaneous liver biopsy. A multicentre retrospective study on 68,276 biopsies. J Hepatol 1986;2:165–73. [DOI] [PubMed] [Google Scholar]
- 11.Johnston DE. Special considerations in interpreting liver function tests. Am Fam Physician 1999;59:2223–30. [PubMed] [Google Scholar]
- 12.Bircher J. Quantitative assessment of deranged hepatic function: a missed opportunity? Semin Liver Dis 1983;3:275–84. [DOI] [PubMed] [Google Scholar]
- 13.McIntyre N. The limitations of conventional liver function tests. Semin Liver Dis 1983;3:265–74. [DOI] [PubMed] [Google Scholar]
- 14.Corbin I, Buist RJ, Peeling J, et al. Utility of hepatic 31-phosphorus magnetic resonance spectroscopy in a rat model of acute liver failure. J Investig Med 2003;51:42–9. [DOI] [PubMed] [Google Scholar]
- 15.Corbin I, Buist RJ, Peeling J, et al. Regenerative activity and liver function following partial hepatectomy in the rat using 31P-MR spectroscopy. Hepatology 2002;36:345–53. [DOI] [PubMed] [Google Scholar]
- 16.Munakata T, Griffiths RD, Martin PA, et al. An in vivo 31P MRS study of patients with liver cirrhosis: progress towards a non-invasive assessment of disease severity. NMR Biomed 1993;6:168–72. [DOI] [PubMed] [Google Scholar]
- 17.Taylor-Robinson SD, Thomas EL, Sargentoni J, et al. Cirrhosis of the human liver: an in vitro 31P nuclear magnetic resonance study. Biochim Biophys Acta 1995;1272:113–18. [DOI] [PubMed] [Google Scholar]
- 18.Taylor-Robinson SD, Sargentoni J, Bell JD, et al. In vivo and in vitro hepatic 31P magnetic resonance spectroscopy and electron microscopy of the cirrhotic liver. Liver 1997;17:198–209. [DOI] [PubMed] [Google Scholar]
- 19.Jeong DH, Jang JJ, Lee SJ, et al. Expression patterns of cell cycle-related proteins in a rat cirrhotic model induced by CCl4 or thioacetamide. J Gastroenterol 2001;36:24–32. [DOI] [PubMed] [Google Scholar]
- 20.Bachmann R, Kreft B, Dombrowski F, et al. Enhanced tumor detection in the presence of liver cirrhosis: experimental study on the diagnostic value of a superparamagnetic iron oxide MR imaging contrast agent (NSR 0430). J Magn Reson Imaging 1999;9:251–6. [DOI] [PubMed] [Google Scholar]
- 21.Rozga J, Foss A, Alumets J, et al. Liver cirrhosis in rats: regeneration and assessment of the role of phenobarbital. J Surg Res 1991;51:329–35. [DOI] [PubMed] [Google Scholar]
- 22.Dupin S, Delrat P, Le Quellec A, et al. Indocyanine green pharmacokinetics in rats with progressive carbon tetrachloride-induced hepatocellular insufficiency. Arzneimittelforschung 1994;44:367–70. [PubMed] [Google Scholar]
- 23.Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol 1984;65:305–11. [PMC free article] [PubMed] [Google Scholar]
- 24.Madhu B, Lagerwall K, Soussi B. Phosphorus metabolites in different muscles of the rat leg by 31P image- selected in vivo spectroscopy. NMR Biomed 1996;9:327–32. [DOI] [PubMed] [Google Scholar]
- 25.Buchli R, Meier D, Martin E, et al. Assessment of absolute metabolite concentrations in human tissue by 31P MRS in vivo. Part II: Muscle, liver, kidney. Magn Reson Med 1994;32:453–8. [DOI] [PubMed] [Google Scholar]
- 26.Meyerhoff DJ, Karczmar GS, Matson GB, et al. Non-invasive quantitation of human liver metabolites using image-guided 31P magnetic resonance spectroscopy. NMR Biomed 1990;3:17–22. [DOI] [PubMed] [Google Scholar]
- 27.Roth K, Hubesch B, Meyerhoff D, et al. Non-invasive quantitation of phosphorus metabolites in human tissue by NMR spectroscopy. J Magn Reson 1989;81:299–310. [Google Scholar]
- 28.Evelhoch JL, Ewy CS, Siegfried BA, et al. 31P spin-lattice relaxation times and resonance linewidths of rat tissue in vivo: dependence upon the static magnetic field strength. Magn Reson Med 1985;2:410–7. [DOI] [PubMed] [Google Scholar]
- 29.Iles RA, Stevens AN, Griffiths JR, et al. Phosphorylation status of liver by 31P-n.m.r. spectroscopy, and its implications for metabolic control. A comparison of 31P-n.m.r. spectroscopy (in vivo and in vitro) with chemical and enzymic determinations of ATP, ADP and Pi. Biochem J 1985;229:141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dashti H, Jeppsson B, Hagerstrand I, et al. Thioacetamide- and carbon tetrachloride- induced liver cirrhosis. Eur Surg Res 1989;21:83–91. [DOI] [PubMed] [Google Scholar]
- 31.Zimmermann T, Muller A, Machnik G, et al. Biochemical and morphological studies on production and regression of experimental liver cirrhosis induced by thioacetamide in Uje: WIST rats. Z Versuchstierkd 1987;30:165–80. [PubMed] [Google Scholar]
- 32.Rosser BG, Gores GJ. Liver cell necrosis: cellular mechanisms and clinical implications. Gastroenterology 1995;108:252–75. [DOI] [PubMed] [Google Scholar]
- 33.Kaita KD, Pettigrew N, Minuk GY. Hepatic regeneration in humans with various liver disease as assessed by Ki-67 staining of formalin-fixed paraffin-embedded liver tissue. Liver 1997;17:13–6. [DOI] [PubMed] [Google Scholar]
- 34.Kawakita N, Seki S, Yanai A, et al. Immunocytochemical identification of proliferative hepatocytes using monoclonal antibody to proliferating cell nuclear antigen (PCNA/cyclin). Comparison with immunocytochemical staining for DNA polymerase-alpha. Am J Clin Pathol 1992;97(suppl 1):S14–20. [PubMed] [Google Scholar]
- 35.Moser M, Zhang M, Gong Y, et al. Effect of preoperative interventions on outcome following liver resection in a rat model of cirrhosis. J Hepatol 2000;32:287–92. [DOI] [PubMed] [Google Scholar]
- 36.Schneeweiss B, Graninger W, Ferenci P, et al. Energy metabolism in patients with acute and chronic liver disease. Hepatology 1990;11:387–93. [DOI] [PubMed] [Google Scholar]
- 37.Muller A, Machnik F, Zimmermann T, et al. Thioacetamide-induced cirrhosis-like liver lesions in rats—usefulness and reliability of this animal model. Exp Pathol 1988;34:229–36. [DOI] [PubMed] [Google Scholar]
- 38.Harvey PJ, Gready JE, Hickey HM, et al. 31P and 1H NMR spectroscopic studies of liver extracts of carbon tetrachloride-treated rats. NMR Biomed 1999;12:395–401. [DOI] [PubMed] [Google Scholar]
- 39.Morgan DJ, McLean AJ. Clinical pharmacokinetic and pharmacodynamic considerations in patients with liver disease. An update. Clin Pharmacokinet 1995;29:370–91. [DOI] [PubMed] [Google Scholar]
- 40.McLean AJ, Morgan DJ. Clinical pharmacokinetics in patients with liver disease. Clin Pharmacokinet 1991;21:42–69. [DOI] [PubMed] [Google Scholar]
- 41.Harvey PJ, Gready JE, Yin Z, et al. Acute oxygen supplementation restores markers of hepatocyte energy status and hypoxia in cirrhotic rats. J Pharmacol Exp Ther 2000;293:641–5. [PubMed] [Google Scholar]
- 42.Katz NR. Metabolic heterogeneity of hepatocytes across the liver acinus. J Nutr 1992;122(suppl):843–9. [DOI] [PubMed] [Google Scholar]
- 43.Gumucio JJ, Miller DL. Functional implications of liver cell heterogeneity. Gastroenterology 1981;80:393–403. [PubMed] [Google Scholar]
- 44.Jungermann K, Katz N. Functional hepatocellular heterogeneity. Hepatology 1982;2:385–95. [DOI] [PubMed] [Google Scholar]
- 45.Seifter S, Englard S. Energy Metabolism, 3rd edn. New York: Raven Press Ltd, 1994.
- 46.Campbell KA, Wu YP, Chacko VP, et al. In vivo 31P NMR spectroscopic changes during liver regeneration. J Surg Res 1990;49:244–7. [DOI] [PubMed] [Google Scholar]
- 47.Morikawa S, Inubushi T, Kitoh K, et al. Chemical assessment of phospholipid and phosphoenergetic metabolites in regenerating rat liver measured by in vivo and in vitro 31P-NMR. Biochim Biophys Acta 1992;1117:251–7. [PubMed] [Google Scholar]
- 48.Murphy EJ, Bates TE, Williams SR, et al. Endoplasmic reticulum: the major contributor to the PDE peak in hepatic 31P-NMR spectra at low magnetic field strengths. Biochim Biophys Acta 1992;1111:51–8. [DOI] [PubMed] [Google Scholar]
- 49.Murphy EJ, Rajagopalan B, Brindle KM, et al. Phospholipid bilayer contribution to 31P NMR spectra in vivo. Magn Reson Med 1989;12:282–9. [DOI] [PubMed] [Google Scholar]
- 50.Ruiz-Cabello J, Cohen JS. Phospholipid metabolites as indicators of cancer cell function. NMR Biomed 1992;5:226–33. [DOI] [PubMed] [Google Scholar]
- 51.Menon DK, Sargentoni J, Taylor-Robinson SD, et al. Effect of functional grade and etiology on in vivo hepatic phosphorus- 31 magnetic resonance spectroscopy in cirrhosis: biochemical basis of spectral appearances. Hepatology 1995;21:417–27. [PubMed] [Google Scholar]