

Abstract
Background and aims: Substance P (SP) release from sensory nerves induces neurogenic inflammation. Neutral endopeptidase (NEP) degrades SP, thereby limiting its proinflammatory effects. Intestinal inflammation following Trichinella spiralis infection markedly downregulates NEP, resulting in diminished SP degradation, with unknown functional consequences. We hypothesised that diminished expression of NEP would exacerbate T spiralis induced enteritis.
Methods: NEP knockout (NEP−/−) and wild-type (NEP+/+) mice were infected with T spiralis and studied at 6, 12, 24, and 48 hours post infection (PI). Tissue inflammation was quantified by computerised cell counting and myeloperoxidase activity (MPO). The leucocyte adhesion molecule, intercellular adhesion molecule 1 (ICAM-1), and SP were assessed by immunohistochemistry.
Results: Before infection, the lack of NEP was not associated with changes in mucosal cellularity or MPO activity. Twelve hours PI, NEP−/− mice showed a 2.5-fold increase in MPO activity at a time when values in NEP+/+ mice were still within normal limits. MPO activity and cellularity peaked at 24 hours PI. This was accompanied by increased staining for both ICAM-1 and SP in NEP−/− mice. Infusion of rhNEP to NEP−/− mice significantly reduced MPO activity 24 hours PI.
Conclusions: These findings demonstrate that NEP downregulates the early onset of nematode intestinal inflammation and that increased bioavailability of SP and overexpression of ICAM-1 in NEP−/− mice likely play a role in the earlier onset of intestinal inflammation.
Keywords: enteric nervous system, inflammation, leucocytes, nematode, tachykinin, endopeptidase
The tachykinin substance P (SP) is a neuropeptide contained in extrinsic (sensory) and intrinsic (enteric) neurones innervating the gastrointestinal tract.1 SP contained in sensory nerves has a dual afferent and efferent function. Its release in the dorsal horn of the spinal cord is crucial for transmission of sensory information from the gut to the central nervous system while its antidromic release regulates several gut functions, including motility, secretion/absorption, and blood flow. Moreover, SP has many proinflammatory effects and abundant evidence indicates that its uncontrolled release within intestinal tissues promotes a sequence of events referred to as “neurogenic inflammation”, which is characterised by arteriolar vasodilation and extravasation of plasma proteins and neutrophils from post capillary venules.2 SP is increased in the inflamed colon of patients with ulcerative colitis,3 pouchitis,4 in the jejunum of Trichinella spiralis infected rats,5 and in other animal models of intestinal inflammation.6,7 Many aspects of neurogenic inflammation are mediated by the interaction of SP with its preferred receptor neurokinin 1 (NK1). In the intestine of patients with inflammatory bowel disease, NK1 receptors are markedly upregulated.8,9 Furthermore, NK1 inactivation, either by pharmacological antagonism or genetic deletion, markedly reduces intestinal inflammation in animal models.10,11 Neurogenic inflammation is terminated by the cell surface enzyme neutral endopeptidase (NEP, EC 3.4.24.11) which degrades SP in the extracellular fluid, thereby terminating its proinflammatory effects.12–16
Little is known of the regulation of NEP expression. Downregulation of NEP would be expected to reduce degradation of SP and other proinflammatory peptides, such as bradykinin (BK), and thereby exacerbate neurogenic inflammation. Consistent with this possibility, we have shown that deletion of NEP markedly enhances colonic inflammation induced by intrarectal instillation of dinitrobenzene sulphonic acid.6 We have previously reported that NEP is markedly downregulated in intestinal mucosa and musculature of rats infected with T spiralis, resulting in a sixfold reduction in the rate of SP degradation.17 These observation raise the possibility that downregulation of NEP in the inflamed intestine could amplify neurogenic inflammation.
Adhesion of leucocytes to endothelial cells and their infiltration into tissues is one of the most critical early events of inflammation.18,19 Leucocyte migration is primarily mediated by the interaction between intercellular adhesion molecule 1 (ICAM)-1 expressed on the endothelium with its ligands β2 integrins (CD11a/CD18 and CD11b/CD18) expressed by leucocytes.20,21 ICAM-1 expression on endothelial cells has been implicated in the early recruitment of leucocytes following intestinal ischaemia and reperfusion,22 as well as surgical manipulation.23 SP interacts with the NK1 receptor to initiate ICAM-1 expression and leucocyte infiltration,24 but the role of NEP in terminating this process is not fully defined. The purpose of the present investigation was to evaluate the role of NEP in regulating the early stages of intestinal inflammation, in particular expression of ICAM-1. To do this, we compared wild-type (NEP+/+) and knockout (NEP−/−) mice. Our aims were to: (1) determine the contribution of NEP to degradation of SP and BK in the intestine; (2) examine whether deletion of NEP results in spontaneous intestinal inflammation; and (3) determine whether deletion of NEP amplifies the initiating events of neurogenic inflammation of the intestine in animals infected with T spiralis.
MATERIALS AND METHODS
Animals
NEP−/− mice back crossed to the C57Bl/6 background were generated as previously described.25 Wild-type littermates and C57Bl/6 mice (Taconic Farms Germantown, New York, USA) of the same background as the NEP−/− mice were used interchangeably as controls. Mice were maintained in a specific pathogen free facility, under controlled ambient temperature and light-dark cycle (14:10 hours), and males and females aged 10–20 weeks were used. All experimental procedures were in accordance with guidelines drafted by the institutional university animal care committees.
Degradation of SP and BK
An extensively washed membrane fraction was prepared from the entire small intestines of three NEP+/+ and three NEP−/− mice.17 Membranes (1 μg protein) were incubated with SP or BK (5 nmol) in 200 µl of 50 mM Tris-HCl, pH 7.4, for 0–120 minutes (found to be optimal in preliminary experiments) at 37°C. Membranes were omitted from controls. When used, 1 µM thiorphan was preincubated with membranes for 10 minutes before addition of substrate. After incubation, samples were boiled for five minutes, centrifuged (14 000 g, five minutes), and the supernatant was acidified with two volumes of 0.5% trifluoroacetic acid in water. Samples were analysed by reversed phase high pressure liquid chromatography.
Extravasation of Evans blue
Mice were anaesthetised with xylazine (10 mg/kg intramuscularly) and ketamine (100 mg/kg intramuscularly). Evans blue (30 mg/kg) in 0.9% saline was injected into a femoral vein. SP (1 nmol/kg intravenously), BK (1 nmol/kg intravenously), or saline (basal conditions) was injected two minutes after Evans blue. When used, thiorphan (2.5 mg/kg intravenously), phosphoramidon (2.5 mg/kg intravenously), or SR 140333 (1 µmol/kg intravenously) was injected five minutes before Evans blue. HOE 140 (0.1 nmol/kg intravenously) was injected 15 minutes before and recombinant human NEP (rhNRP 3 mg/kg intravenously) 10 minutes before Evans blue. All drugs and recombinant NEP were dissolved in 0.9% saline, with the exception of SR 140333 which was dissolved in dimethyl sulphoxide (dimethyl sulphoxide had no effect on basal extravasation). Ten minutes after injection of Evans blue, mice were transcardially perfused with 70 ml of 100 mM phosphate buffered saline (PBS), pH 7.4, containing 100 units/ml heparin, and 200 ml of 4% paraformaldehyde in PBS. The jejunum was removed, rinsed in saline, gently blotted, and weighed. Half of each sample was dried at 60°C for 48 hours, and half was placed in 1 ml formamide for 48 hours to extract Evans blue. Evans blue was quantified by measuring absorption at 620 nm, and absorption was compared with a standard curve.
Infection with T spiralis
Mice were infected by intragastric inoculation of 300–400 T spiralis larvae in 0.1 ml PBS. The larvae were obtained from infected CD1 mice, as previously described.26
Histology and immunohistochemistry
Full thickness samples of jejunum were rapidly dissected and processed for either haematoxylin-eosin histology or immunohistochemistry. For the latter, tissues were fixed by immersion in 4% paraformaldehyde in 0.1 M PBS for 6–8 hours at 4°C and subsequently placed in 25% sucrose in 0.1 M PBS for cryoprotection until sectioning. Tissue specimens were cut with a cryostat at 10–12 μm, mounted onto chome-alum gelatin coated slides, and stored at −30°C until processed for immunohistochemistry. Tissue sections were processed with the avidin-biotin-peroxidase complex method, as previously described.27,28 Briefly, section were washed in 0.1 M PBS, pretreated for 30 minutes at room temperature with 10% normal goat serum, and incubated with a rabbit polyclonal antibody directed against the C terminal portion of the SP peptide (8701; working dilution 1:5000)27,28 or with a rat monoclonal antibody recognising the mouse ICAM-1 molecule (R&D Systems Minneapolis, Minnesota, USA), overnight at 4°C in a humid chamber. Sections were then washed in PBS and incubated for two hours at room temperature in affinity purified goat antirabbit or antirat biotinylated IgG (dilution 1:100) (Vector Laboratories, Burlingame, California, USA), followed by avidin-biotin-peroxidase solution for 30 minutes, and then exposed to 3,3′-diaminobenzidine with 0.01% H2O2. Sections were finally dehydrated and coverslipped with mounting medium. To reduce non-specific staining due to endogenous peroxidase, tissue sections were first dehydrated, placed in 100% methanol, and then in a solution composed of 98% methanol, 1% acetic acid, and 1% sodium nitroferricyanide for 15 minutes, followed by 100% methanol, rehydrated, and then incubated with the primary antibody. Both primary and secondary antibodies were diluted in 0.5 % Triton-X-100 in 0.1 M PBS. Sections were analysed with a Leitz Dialux microscope using bright field optics. Specificity studies included:27,28 (a) omission of the primary antibody; (b) substitution of the primary antibody with normal rabbit, mouse, or rat sera (dilution of 1:50), and (c) incubation with the primary antibody preadsorbed for 12–16 hours at 4°C with synthetic homologous peptides (Bachem, Torrance, California, USA; R&D Systems) including SP, neurokinin A, neurokinin B, and ICAM-1 (10 μmol/l).
Mucosal and submucosal cell counts
Lamina propria and submucosal cells were quantified on haematoxylin-eosin cross sections with a Leitz Dialux microscope (×25 objective) by two operators (GB and CC) in a blinded fashion using a computer assisted analysis system (Cytometrica software; C&V, Bologna, Italy). All measurements were performed using a modification of previously published methods.29–31 Briefly, the fields in which the plane of the section was well oriented, with crypts and villi perpendicular to the muscularis propria, were digitised and randomly sampled with the aid of a grid (0.5 mm2) located below the slide. A stereological grid with cross shaped points was overlaid to the digitised sampled fields by computer software and used to determine the relative area of cellular nuclei located within the lamina propria and the submucosa. To obtain the relative area occupied by cell nuclei in the microscopic field, the number of points hitting cell nuclei was divided by the total number of points in the frame.
Myeloperoxidase activity
Samples of jejunum (50–100 mg) were obtained for assessment of myeloperoxidase (MPO), snap frozen in liquid nitrogen, and assayed within seven days using a previously described method.32 MPO was expressed as U/mg of tissue where 1 U corresponds to the activity of enzyme required to degrade 1 µmol of hydrogen peroxide in one minute at 24°C. MPO is a granule associated enzyme primarily contained in neutrophils and other cells of myeloid origin and is widely used as a marker of intestinal inflammation.33
Drugs and solutions
Unless otherwise stated, all reagents were obtained from Sigma Chemical Co (St Louis, Missouri, USA). rhNEP (Khepri Pharmaceuticals, South San Francisco, California, USA) (3 mg/kg/day) or bovine serum albumin (BSA) (control, 3 mg/kg/day) were dissolved in saline and infused subcutaneously from osmotic minipumps implanted beneath the back skin under Enfluorane anaesthesia, 24 hours before T spiralis infection up to sacrifice.
Data expression and statistical analysis
Results are expressed as mean (SEM) (n>4 mice per group). For mucosal cellularity, results are expressed as relative area (%) occupied by cellular nuclei in the lamina propria and submucosa. Statistical significance was determined at p<0.05 using the Student’s t test for comparison of two means or one way analysis of variance for comparison of more than two means, followed by post hoc tests using Bonferroni’s correction.
RESULTS
Degradation of SP and BK
We compared degradation of SP and BK by membranes prepared from the intestine of NEP+/+ and NEP−/− mice to determine the importance of NEP in peptide degradation. Membranes prepared from the small intestine of NEP+/+ mice rapidly degraded SP and BK (fig 1A, B ▶). The NEP inhibitor thiorphan reduced degradation of SP by 43% and BK by 70% (determined after 120 minute incubation period of peptides with membranes) (fig 1B ▶). Membranes from NEP−/− mice degraded SP 43% more slowly and BK 33% more slowly than membranes from NEP+/+ mice (determined after 120 minute incubation period of peptides with membranes) (fig 1B ▶). These results show that NEP plays a major role in the degradation of SP and BK by membrane associated proteases in the intestine. The observation that peptide degradation is markedly diminished in NEP−/− mice suggests that there is no compensatory upregulation of other proteases.
Figure 1.

Degradation of substance P (SP) and bradykinin (BK) by intestinal membranes from wild-type (NEP+/+) and knockout (NEP−/−) neutral endopeptidase mice. (A) High pressure liquid chromatograms of the degradation products after 120 minute incubation period with or without thiorphan. (B) Time course of degradation of SP and BK with or without thiorphan, presented as per cent degradation compared with controls in which membranes were omitted. In NEP+/+ mice, SP and BK were rapidly degraded and the NEP inhibitor thiorphan reduced degradation of SP by 43% and BK by 70% (determined after 120 minute incubation period of peptides with membranes). Membranes from NEP−/− mice degraded SP 43% more slowly and BK 33% more slowly than membranes from NEP+/+ mice (determined after 120 minute incubation period of peptides with membranes). The results in (A) are representative chromatograms from 4–6 analyses. The results in (B) are the mean of n = 2 or 3 experiments, each in duplicate. OD, optical density.
Plasma extravasation
We compared plasma extravasation in NEP+/+ and NEP−/− mice under basal and stimulated conditions to determine if the absence of NEP resulted in spontaneous neurogenic inflammation. In NEP+/+ mice, SP and BK stimulated extravasation of Evans blue in the jejunum by >3-fold over basal (fig 2A ▶). The response to SP was abolished by the NK1 receptor antagonist SR140333 but unaffected by the B2R antagonist HOE140, indicating involvement of the NK1 receptor. The response to BK was abolished by HOE140, indicating that BK stimulates plasma extravasation by activating B2R. BK stimulated extravasation of Evans blue was also inhibited by SR140333, suggesting that the response is partially dependent on release of SP and activation of the NK1 receptor.
Figure 2.

Extravasation of Evans blue in the jejunum. (A) Basal, substance P (SP), and bradykinin (BK) stimulated extravasation of Evans blue in wild-type (NEP+/+) and knockout (NEP−/−) neutral endopeptidase mice (mean (SEM) (n)). *p<0.05 compared with basal (ANOVA, Bonferroni t test). (B) Effects of thiorphan (Thio) and phosphoramidon (Phos) on basal extravasation of Evans blue in NEP+/+ mice, and effects of human recombinant NEP (rhNEP), SR140333, and HOE140 on basal extravasation in NEP−/− mice (mean (SEM) (n)). *p<0.05 compared with basal for wild-type mice; †p<0.05 compared with basal for NEP knockout mice (ANOVA, Bonferroni t test).
Under basal conditions, extravasation of Evans blue in the jejunum of NEP−/− mice was fourfold greater than in NEP+/+ mice (fig 2A, B ▶). The basal leak was so high that exogenous SP and BK failed to further stimulate plasma extravasation in NEP−/− animals. This elevated basal extravasation was prevented by prior administration of rhNEP, and was markedly inhibited by administration of SR140333 or HOE140 (fig 2B ▶). Conversely, administration of the NEP inhibitors phosphoramidon or thiorphan to NEP+/+ mice stimulated Evans blue extravasation to levels observed under basal conditions in NEP−/− mice. Together, these results indicate that NEP deletion causes elevated plasma extravasation in the jejunum. This event is due to diminished degradation of SP and BK with consequent activation of NK1 receptor and B2R.
Mucosal and submucosal cell counts
NEP deletion diminished SP and BK degradation resulting in spontaneous plasma extravasation. These findings raised the possibility that NEP−/− mice would be highly sensitive to inflammatory stimuli. In the next experiments we examined this possibility by comparing T spiralis induced inflammation in NEP+/+ and NEP−/− mice.
As shown in fig 3 ▶, before T spiralis infection, lamina propria and submucosal cellularity from NEP−/− mice was not statistically different. After infection, NEP−/− mice showed a significant increase in lamina propria and submucosal cellularity over non-infected NEP−/− mice, which was detectable by 24 hours or 48 hours PI (p<0.05) at a time when the cellularity in NEP+/+ mice was not statistically different from controls. At 48 hours PI, lamina propria and submucosal cellularity in NEP−/− mice was significantly increased over values obtained at the corresponding time points PI in NEP+/+ mice (p<0.05).
Figure 3.
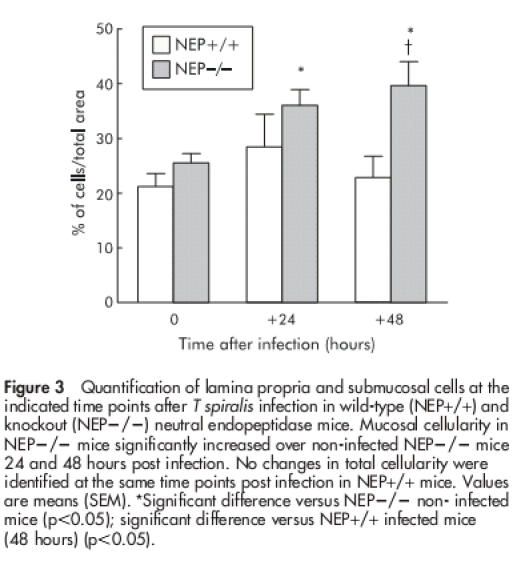
Quantification of lamina propria and submucosal cells at the indicated time points after T spiralis infection in wild-type (NEP+/+) and knockout (NEP−/−) neutral endopeptidase mice. Mucosal cellularity in NEP−/− mice significantly increased over non-infected NEP−/− mice 24 and 48 hours post infection. No changes in total cellularity were identified at the same time points post infection in NEP+/+ mice. Values are means (SEM). *Significant difference versus NEP−/− non- infected mice (p<0.05); significant difference versus NEP+/+ infected mice (48 hours) (p<0.05).
MPO activity
As shown in fig 4 ▶, before T spiralis infection, MPO activity in jejunal extracts from NEP+/+ and NEP−/− mice was the same. In NEP+/+ mice, a significant increase in MPO activity over non-infected controls was detectable by 24 hours PI. In contrast, MPO activity in NEP−/− mice increased as early as 12 hours PI. At 12, 24, and 48 hours PI, MPO activity in NEP−/− was increased by about 2.5-fold over values obtained at the corresponding time points PI in NEP+/+ mice (p<0.01).
Figure 4.

Myeloperoxidase (MPO) activity from the jejunum at the indicated time points after T spiralis infection in wild-type (NEP+/+) and knockout (NEP−/−) neutral endopeptidase mice. After infection, MPO activity increased significantly earlier (12 hours) and with a higher magnitude in NEP−/− mice in comparison with NEP+/+ mice. Values are means (SEM). **Significant difference versus non-infected mice (p<0.01); ††significant difference versus NEP+/+ studied at the same time post infection (p<0.01).
Prevention of intestinal inflammation by administration of rhNEP
To determine whether the increased MPO activity PI in NEP−/− mice could be attenuated by exogenous administration of NEP, mice were treated with rhNEP 24 hours before and for 24 hours after T spiralis infection. As shown in fig 5 ▶, rhNEP significantly attenuated the increased MPO activity in NEP−/− infected mice compared with NEP−/− infected mice receiving BSA (control). rhNEP had no effect on MPO activity from NEP−/− non-infected mice (fig 5 ▶).
Figure 5.
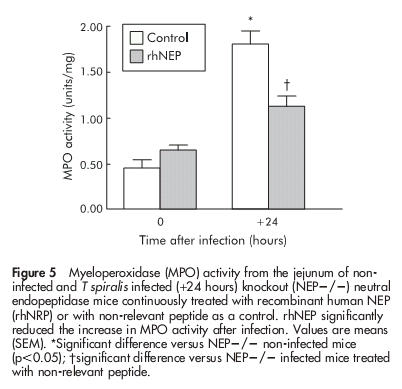
Myeloperoxidase (MPO) activity from the jejunum of non-infected and T spiralis infected (+24 hours) knockout (NEP−/−) neutral endopeptidase mice continuously treated with recombinant human NEP (rhNRP) or with non-relevant peptide as a control. rhNEP significantly reduced the increase in MPO activity after infection. Values are means (SEM). *Significant difference versus NEP−/− non-infected mice (p<0.05); †significant difference versus NEP−/− infected mice treated with non-relevant peptide.
Substance P and ICAM-1 immunoreactivity
As shown in fig 6 ▶, in non-infected NEP−/− or NEP+/+ mice, SP immunolabelling was detected in varicose nerve endings targeting the different layers of the gut wall as well as in myenteric and submucosal ganglion cell bodies (fig 6A, B ▶). Before infection, the distribution pattern of SP immunoreactivity as well as its staining intensity in NEP−/− was not different from that observed in NEP+/+ mice (fig 6A, B ▶). At 24 hours PI, there was an increase in SP staining intensity in NEP+/+ mice (fig 6C ▶). However, at the same time point PI, a much more evident increase in SP immunoreactive neural network and staining intensity was observed in NEP−/− compared with NEP+/+ mice (fig 6D ▶).
Figure 6.

Representative photomicrographs of jejunum showing substance P (SP) immunoreactivity in neuronal cell bodies and nerve processes before and after T spiralis infection. Under basal conditions, SP immunoreactive neural pattern and staining intensity was comparable in wild-type (NEP+/+) (A) and knockout (NEP−/−) (B) neutral endopeptidase mice. T spiralis infection (24 hours) evoked an increase in SP staining intensity in NEP+/+ mice (C). However, the increase in both SP density and staining intensity after infection in NEP−/− mice (D) was markedly enhanced. Calibration bar 25 µm.
We next examined the effect of NEP deletion on ICAM-1 immunoreactivity in the intestine. Before infection, NEP+/+ mice showed a weak ICAM-1 immunoreactivity which was observed in the submucosal and mucosal layer (fig 7A ▶). This ICAM-1 immunoreactive pattern showed a consistent increase in non-infected NEP−/− mice (fig 7B ▶). Following T spiralis infection (24 hours), there was no evident increase in ICAM-1 immunoreactivity in NEP+/+ mice (fig 7C ▶). However, at the same time point PI, NEP−/− showed a markedly enhanced ICAM-1 immunoreactivity both at the submucosa and mucosal levels (fig 7D ▶).
Figure 7.

Representative photomicrographs showing intercellular adhesion molecule 1 (ICAM-1) immunoreactive pattern throughout the jejunal wall before and after T spiralis infection. Under basal conditions, ICAM-1 was weakly expressed in the submucosal and mucosal layer in wild-type (NEP+/+) neutral endopeptidase mice (A), this pattern showed a detectable increase in knockout (NEP−/−) mice (B). Following T spiralis infection (24 hours), no evident increase in ICAM-1 immunoreactivity was appreciated in NEP+/+ infected mice (C). However, NEP−/− infected (24 hours) mice showed markedly enhanced ICAM-1 immunoreactivity both at the submucosal and mucosal levels (D). Calibration bar 25 μm.
DISCUSSION
In this study, we determined the role of NEP in the initial steps of intestinal inflammation caused by T spiralis infection. We correlated intestinal infiltration of leucocytes with expression of the proinflammatory neuropeptide SP, which is inactivated by NEP, and the adhesion molecule ICAM-1, which is required for leucocyte recruitment. Our results demonstrated that the lack of NEP was associated with an earlier onset and an increased magnitude of intestinal inflammation following T spiralis infection. These changes were accompanied by enhanced expression of SP and ICAM-1.
Our results showed that NEP deletion markedly diminished degradation of SP and BK in the intestine and caused spontaneous plasma extravasation in the jejunum that was a functional consequence of this diminished degradation. Our results are supported by previous studies in NEP−/− mice demonstrating that under basal conditions these animals display increased extravasation of plasma in the duodenum16 as well as in the colon6 without evidence of overt inflammation at the tissue level.6 In line with these results, we found comparable values for mucosal cell counts and MPO activity from the jejunum of non-infected NEP−/− and NEP+/+ mice. Taken together, these results suggest that the lack of NEP is not sufficient to cause an overt spontaneous intestinal inflammation throughout the small and large intestine. Following T spiralis infection however we found that intestinal inflammatory infiltration was markedly increased in NEP−/− mice compared with the gene bearing animals. This increased mucosal cellularity in NEP−/− mice was paralleled by a significant 2.5-fold increase in MPO activity which reflected a marked granulocyte infiltration (that is, neutrophils and eosinophils) into the tissue. Exogenous administration of rhNEP to NEP−/− infected mice markedly reduced MPO activity, thus confirming that the exaggerated post infectious inflammatory response observed in NEP−/− mice was mediated by an NEP substrate. Our data demonstrated that, after infection, infiltrating leucocytes could be detected significantly earlier (12 hours) in NEP−/− mice compared with the gene bearing mice (24 hours). These data suggest that NEP plays a crucial role in priming intestinal recruitment of leucocytes in the early stage of infection. In addition, these findings, taken in conjunction with our previous demonstration that NEP terminates colonic inflammation induced by dinitrobenzene sulphonic acid,6 show that NEP acts throughout the small and large intestine to protect uncontrolled intestinal inflammation induced by different noxious stimuli.
Previous studies by this laboratory in the rat showed that T spiralis infection evokes a marked increase in SP immunoreactivity in the enteric neural network5 and particularly in sensory afferent fibres projecting to the intestine.34 Moreover, there is a marked downregulation of NEP and diminished degradation of SP in infected animals.17 The results from the present study are in keeping with these previous data and also demonstrate that the lack of NEP further amplifies the increase in SP immunoreactivity in enteric nerves after T spiralis infection. It is likely that this increase in SP can be ascribed to a lack of degradation of this neuropeptide by NEP12,13 but other mechanisms cannot be discarded. For example, excessive SP release from sensory nerves could be evoked by another peptide, namely BK, which is also degraded by NEP.35
SP has a wide array of proinflammatory effects, including gap formation between endothelial cells of postcapillary venules to induce plasma protein extravasation, inflammatory cell recruitment, and degranulation of intestinal mast cells.2,24,36,37 Our demonstration of increased MPO activity and mucosal cellularity in NEP−/− infected mice suggests that SP is involved in the initiation of tissue leucocyte recruitment.2 In keeping with this hypothesis, Castagliuolo and colleagues7 demonstrated that application of Clostridium difficile toxin A to the rat intestinal lumen is accompanied by rapid SP mRNA expression and protein increase which occurs prior to changes in fluid secretion, mucosal permeability, and histological evidence of enteritis. Furthermore, pharmacological blockade of NK1 receptor markedly reduced colonic inflammation in dinitrobenzene sulphonic acid treated NEP−/− mice.6
The interaction between adhesion molecules expressed by endothelial cells with their ligands on circulating leucocytes have been recently shown to be of crucial importance in nematode induced intestinal inflammation and in the development of a Th2 type immune response.38,39 In the present study we found that expression of the adhesion molecule ICAM-1 was markedly enhanced in NEP−/− mice after T spiralis infection. This increase may explain the exaggerated leucocyte intestinal infiltration in NEP−/− mice after infection. Although the mechanisms underlying increased ICAM-1 expression are unknown, it is likely that SP plays an important role. In fact, previous studies have demonstrated that SP evokes ICAM-1 expression via NK1 activation on endothelial cells which contributes to leucocyte recruitment and tissue injury.24,40
In conclusion, this study showed for the first time that NEP plays a pivotal role in the downregulation of the onset of intestinal inflammation evoked by T spiralis infection. An increased bioavailability of SP and overexpression of the adhesion molecule ICAM-1 may be involved in the increased inflammatory response observed in NEP−/− mice. We postulate that neurogenic mechanisms involving the early release of SP play a pivotal role in the initiation of intestinal inflammation in this model.
Acknowledgments
This work was supported by a grant from the Medical Research Council of Canada (SMC) and by NIH grant DK52388 (EFG, NWB, and SMC), DK39957, DK43207 (NWB). The authors wish to thank Dr P Chieco for his expertise and guidance in quantitative analysis performed on histological specimens.
Abbreviations
ICAM-1, intercellular adhesion molecule 1
MPO, myeloperoxidase
NEP, neutral endopeptidase
rhNRP, recombinant human NEP
NEP+/+, wild-type mice
NEP−/−, knockout mice
NK1, neurokinin 1
PI, post infection
SP, substance P
BK, bradykinin
PBS, phosphate buffered saline
BSA, bovine serum albumin
REFERENCES
- 1.Otsuka M, Yoshioka K. Neurotransmitter functions of mammalian tachykinins. Physiol Rev 1993;73:229–308. [DOI] [PubMed] [Google Scholar]
- 2.McDonald DM, Bowden JJ, Baluk P, et al. Neurogenic inflammation. A model for studying efferent actions of sensory nerves. Adv Exp Med Biol 1996;410:453–62. [PubMed] [Google Scholar]
- 3.Goldin E, Karmeli F, Selinger Z, et al. Colonic substance P levels are increased in ulcerative colitis and decreased in chonic severe constipation. Dig Dis Sci 1989;34:754–57. [DOI] [PubMed] [Google Scholar]
- 4.Keranen U, Jarvinen H, Kiviluoto T, et al. Substance P- and vasoactive intestinal polypeptide-immunoreactive innervation in normal and inflamed pouches after restorative proctocolectomy for ulcerative colitis. Dig Dis Sci 1996;41:1658–64. [DOI] [PubMed] [Google Scholar]
- 5.Swain MG, Agro A, Blennerhassett P, et al. Increased levels of substance P in the myenteric plexus of Trichinella-infected rats. Gastroenterology 1992;102:1913–19. [DOI] [PubMed] [Google Scholar]
- 6.Sturiale S, Barbara G, Qiu B, et al. Neutral endopeptidase (EC 3.4.24.11) terminates colitis by degrading substance P. Proc Natl Acad Sci U S A 1999;96:11653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castagliuolo I, Keates AC, Qiu B, et al. Increased substance P responses in dorsal root ganglia and intestinal macrophages Clostridium difficile toxin enteritis in rats. Proc Natl Acad Sci U S A 1997;94:4788–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantyh CR, Gates TS, Zimmerman RP, et al. Receptor binding sites for substance P, but not substance K or neuromedin K, are expressed in high concentrations by arterioles, venules, and lymph nodules in surgical specimens obtained from patients with ulcerative colitis and Crohn disease. Proc Natl Acad Sci U S A 1988;85:3235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goode T, O’Connel J, Anton P, et al. Neurokinin-1 receptor expression in inflammatory bowel disease: molecular quantitation and localisation. Gut 2000;47:387–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pothoulakis C, Castagliuolo I, LaMont JT, et al. CP-96,345, a substance P antagonist, inhibits rat intestinal responses to Clostridium difficile toxin A but not cholera toxin. Proc Natl Acad Sci U S A 1994;91:947–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castagliuolo I, Riegler M, Pasha A, et al. Neurokinin-1 (NK-1) receptor is required in Clostridium difficile-induced enteritis. J Clin Invest 1998;101:1547–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsas R, Fulcher IS, Kenny AJ, et al. Substance P and (Leu)enkephalin are hydrolyzed by an enzyme in pig caudate synaptic membranes that is identical with the endopeptidase of kidney microvilli. Proc Natl Acad Sci U S A 1983;80:3111–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okamoto A, Lovett M, Payan DG, et al. Interactions between neutral endopeptidase (EC 3.4.24.11) and the substance P (NK 1) receptor expressed in mammalian cells. Biochem J 1994;299:683–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umeno E, Nadel JA, Huang HAT, et al. Inhibition of neutral endopeptidase potentiates neurogenic inflammation in the rat trachea. J Appl Physiol 1989;66:2647–52. [DOI] [PubMed] [Google Scholar]
- 15.Umeno E, Nadel JA, McDonald DM. Neurogenic inflammation of the rat trachea: fate of neutrophils that adhere to venules. J Appl Physiol 1990;69:2131–6. [DOI] [PubMed] [Google Scholar]
- 16.Lu B, Figini M, Emanueli C, et al. The control of microvascular permeability and blood pressure by neutral endopeptidase. Nat Med 1997;3:904–7. [DOI] [PubMed] [Google Scholar]
- 17.Hwang T, Leichter R, Okamoto A, et al. Downregulation of neutral endopeptidase (EC 3.4.24.11) in the inflamed rat intestine. Am J Physiol 1993;264:G735–43. [DOI] [PubMed] [Google Scholar]
- 18.Granger DN. Cell adhesion and migration. II. Leukocyte-endothelial cell adhesion in the digestive system. Am J Physiol 1997;273:G982–6. [DOI] [PubMed] [Google Scholar]
- 19.Granger DN, Kubes P. The microcirculation and inflammation: modulation of leukocyte-endothelial cell adhesion. J Leukoc Biol 1994;55:662–75. [PubMed] [Google Scholar]
- 20.Carlos TM, Harlan JM. Leukocyte-endothelial adhesion molecules. Blood 1994;84:2069–101. [PubMed] [Google Scholar]
- 21.Horie Y, Wolf R, Miyasaka M, et al. Leukocyte adhesion and hepatic microvascular responses to intestinal ischemia/reperfusion in rats. Gastroenterology 1996;111:666–73. [DOI] [PubMed] [Google Scholar]
- 22.Russell J, Epstein CJ, Grisham MB, et al. Regulation of E-selectin expression in postischemic intestinal microvasculature. Am J Physiol Gastrointest Liver Physiol 2000;278:G878–85. [DOI] [PubMed] [Google Scholar]
- 23.Kalff JC, Carlos TM, Schaut WH, et al. Surgically induced leukocytic infiltrates within the rat intestinal muscularis mediate postoperative ileus. Gastroenterology 1999;117:378–87. [DOI] [PubMed] [Google Scholar]
- 24.Quinlan KL, Song IS, Bunnet NW, et al. Neuropeptide regulation of human dermal microvascular endothelial cell ICAM-1 expression and function. Am J Physiol 1998;275:C1580–90. [DOI] [PubMed] [Google Scholar]
- 25.Lu B, Gerard NP, Kolakowski LF jr, et al. Neutral endopeptidase modulation of septic shock. J Exp Med 1995;181:2271–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vermillion DL, Collins SM. Increased responsiveness of jejunal longitudinal muscle in Trichinella-infected rats. Am J Physiol 1988;254:G124–9. [DOI] [PubMed] [Google Scholar]
- 27.De Giorgio R, Sternini C, Anderson K, et al. Tissue distribution and innervation pattern of peptide immunoreactivities in the rat pancreas. Peptides 1992;13:91–8. [DOI] [PubMed] [Google Scholar]
- 28.De Giorgio R, Zittel TT, Parodi JE, et al. Peptide immunoreactivities in the ganglionated plexuses and nerve fibers innervating the human gallbladder. J Autonom Nerv Syst 1995;51:37–47. [DOI] [PubMed] [Google Scholar]
- 29.Chieco P, Jonker A, Melchiorri C, et al. A user’s guide for avoiding errors in absorbance image cytometry: a review with original experimental observations. Histochem J 1994;26:1–19. [PubMed] [Google Scholar]
- 30.Gundersen HJC, Bendtsen TF, Korbo L, et al. Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS 1988;96:379–94. [DOI] [PubMed] [Google Scholar]
- 31.Lee E, Schiller LR, Fordtran JS. Quantification of colonic lamina propria cells by means of a morphometric point-counting method. Gastroenterology 1988;94:409–18. [DOI] [PubMed] [Google Scholar]
- 32.Boughton-Smith NK, Wallace JL, Whittle BJ. Relationship between arachidonic acid metabolism, myeloperoxidase activity and leukocyte infiltration in a rat model of inflammatory bowel disease. Agents Actions 1988;25:115–23. [DOI] [PubMed] [Google Scholar]
- 33.Smith JW, Castro GA. Relation of peroxidase activity in gut mucosa to inflammation. Am J Physiol 1978;235:R72–9. [DOI] [PubMed] [Google Scholar]
- 34.De Giorgio R, Barbara G, Blennerhassett P, et al. Intestinal inflammation and activation of sensory nerve pathways: a functional and morphological study in the nematode infected rat. Gut 2001;49:822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Figini M, Emanueli C, Grady EF, et al. Substance P and bradykinin stimulate plasma extravasation in the mouse gastrointestinal tract and pancreas. Am J Physiol 1997;272:G785–93. [DOI] [PubMed] [Google Scholar]
- 36.Bowden JJ, Garland AM, Baluk P, et al. Direct observation of substance P-induced internalization of neurokinin 1 (NK1) receptors at sites of inflammation. Proc Natl Acad Sci U S A 1994;91:8964–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang L, Stanisz AM, Wershil BK, et al. Substance P induces ion secretion in mouse small intestine though effects on enteric nerves and mast cells. Am J Physiol 1995;269:G85–92. [DOI] [PubMed] [Google Scholar]
- 38.Artis D, Humpheys NE, Potten CS, et al. β7 integrin-deficient mice: delayed leukocyte recruitment and attenuated protective immunity in the small intestine during enteric helminth infection. Eur J Immunol 2000;30:1656–64. [DOI] [PubMed] [Google Scholar]
- 39.Wang Z, Takamoto M, Sugane K. Costimulatory signal though CD86 is important in Th2 response in Trichinella spiralis infected mice. Parasite Immunol 2000;22:121–30. [DOI] [PubMed] [Google Scholar]
- 40.DeRose V, Robbins RA, Snider RM, et al. Substance P increases neutrophil adhesion to bronchial epithelial cells. J Immunol 1994;152:1339–46. [PubMed] [Google Scholar]