

Abstract
Background: Cholera toxin (CT) acts on intestinal epithelial cells both directly and indirectly via activation of a secretory neural reflex. The reflex may release acetylcholine as one of its final neurotransmitters. This opens up the possibility of a third mechanism of action for CT, namely a synergistic interaction between two secretagogues acting on different second messenger systems within the epithelial cell.
Aims: To establish evidence for cholinergic innervation to human ileal epithelial cells and to investigate whether CT potentiates the action of acetylcholine on human intestinal epithelial cells.
Methods: Transverse sections of human ileum were examined for mucosal cholinergic nerves and M3 muscarinic receptors using antibodies raised to choline acetyltransferase and M3 receptors. Short circuit current (Isc) responses and ion flux movements were elicited from T84 epithelial cell monolayers set up in Ussing chambers.
Results: Immunohistochemistry of native human ileal mucosa revealed the presence of both cholinergic nerves and muscarinic M3 receptors located to the basolateral domain of epithelial cells. Secretory responses of T84 cell monolayers to acetylcholine were greatly potentiated in the presence of CT. This effect, substituting forskolin for CT, was mirrored by increases in basolateral 86Rb and apical 125I efflux. Charybdotoxin plus apamin reduced both Isc and 86Rb efflux evoked by acetylcholine, in the presence of forskolin.
Conclusions: Human ileal mucosa receives a direct cholinergic innervation to its epithelial cells. Secretory effects of acetylcholine on epithelial cells are augmented in the presence of CT. Such a synergistic response is dependent on optimum opening of basolateral potassium channels by acetylcholine and apical chloride channels by CT. The interaction may contribute to the mechanism of action of cholera toxin induced secretory diarrhoea.
Keywords: acetylcholine, cholera toxin, intestinal secretion, T84 cell line, muscarinic receptors, ileal mucosa, cholinergic nerves
Vibrio cholerae is known to cause profuse watery diarrhoea primarily through the action of cholera toxin (CT) on the intestinal mucosa. On isolated intestinal mucosal preparations mounted in Ussing chambers, CT increases cAMP levels and short circuit current (Isc).1 The apparent non-neural nature of this action was confirmed by similar findings using the T84 human colonic cell line2 and by observations that CT elicits secretory responses from human ileal mucosa treated with tetrodotoxin.3 However, both the magnitude of diarrhoea in cholera and the likelihood that access of the bacterium to intestinal crypts may be restricted has prompted investigations into additional mechanisms of action that may amplify the effects of the toxin.
Convincing evidence that CT activates a secretomotor neural reflex arose from observations that CT induced diarrhoea was reduced by drugs such as local anaesthetics, tetrodotoxin, and hexamethonium.4 The reflex required the presence of the myenteric plexus5 and utilised secretomotor neurones, the most prominent of which appear to be those containing vasoactive intestinal polypeptide (VIP) and acetylcholine (ACh).6,7
The major body of evidence supports a role for ACh as a putative neurotransmitter directly involved in epithelial cell electrolyte transport and comes from anatomical, functional, and receptor binding studies.7–9 However, investigations using muscle stripped preparations of rat colon set up in Ussing chambers have demonstrated that ACh also acts indirectly through activation of uncharacterised secretomotor neurones.10,11 Anatomical studies in the guinea pig and pig have demonstrated cholinergic neurones projecting to the intestinal mucosa.7,12,13 However, cholinergic neurones could not be demonstrated innervating mucosa of the human colon or ileum.14,15 This may represent species differences or more likely a false negative (SJ Brookes, personal communication).
In humans therefore, if ACh released by CT exerts a direct action on epithelial cells, then it is possible that there may be a synergistic interaction between the two secretagogues at the level of the enterocyte.16,17
Using native human ileal mucosa and human intestinal epithelial cells (T84), we have investigated whether there is cholinergic innervation to human ileal enterocytes and if there is a synergistic interaction between CT and the neurotransmitter ACh. Positive evidence for both would be clinically significant in representing an additional mechanism whereby diarrhoea would be worsened in cholera.
METHODS
Cell culture
T84 cells, a human colonic epithelial cell line, were obtained from the European Collection of Cell Cultures (ECACC, Salisbury, Wiltshire, UK) and used between passages 70 and 85. The methods have been described previously.18 One million or four million cells were seeded onto 12 mm or 24 mm internal diameter collagenised semipermeable membranes, respectively (Costar Snapwell inserts, 0.4 μM pore diameter) and after nine or 11 days the inserts with attached confluent monolayer were used for experimentation.
Isc measurements
Monolayers on 12 mm inserts were placed into a modified Ussing chamber for continuous recording of Isc. Monolayers were bathed on both sides with 10 ml of circulating gassed Kreb’s buffer kept at 37°C (2.5 ml for isotope experiments). Electrical measurements were carried out as discussed previously.18 CT was applied to the apical domain of monolayers unless indicated otherwise. All other drugs were given basolaterally. No monolayer received more than one concentration of a given compound.
Measurement of ion efflux
We used previously validated methods in T84 cells19 to measure basal and stimulated cellular effluxes of 125I and 86Rb as a measure of the activities of apical chloride and basolateral potassium channels, respectively. Cells were seeded onto 12 mm inserts as described, and cultured until confluent. Inserts and confluent monolayers were loaded for 180 minutes in medium containing either 2.5 μCi/ml 125I or 1 μCi/ml 86Rb. Inserts were rapidly washed following which 500 μl aliquots of radioactive Kreb’s were removed and replaced with the same volume of isotope free Kreb’s sequentially at two minute intervals for a 22 minute period from either the apical or basolateral compartments for 125I and 86Rb experiments, respectively. At the end of the experiments, isotope remaining in the monolayer was extracted with 0.1 M NaOH and the extract solution counted with the aliquot samples. For the efflux experiments, data were corrected for sampling volume (0.5 ml from a 2.5 ml reservoir) and previous sampling loss due to buffer replacement. Efflux rate constants were calculated as described by Venglarik and colleagues.19 Briefly, release of isotope from monolayers (efflux) was calculated as a percentage of the total (tissue and solution) radioactivity present at the beginning of a collection period. Rate constants (r) were calculated by the following equation:
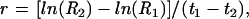 |
where R1 and R2 are the percentages of counts released in the cell layer at times 1 (t1) and 2 (t2), respectively.19 Experiments were performed with forskolin as a cAMP dependent agonist, because it was not technically possible to use CT within the time frame of the experiments, due to the slow onset of action of the toxin.
cAMP measurements
Cells were cultured as previously described and then seeded onto large 24 mm diameter inserts. When confluent, the inserts were rapidly washed and bathed in pregassed Kreb’s at 37°C in a 5% CO2 atmosphere. 3-Isobutyl-1-methylxanthine (IBMX) 0.1 mM was added to Kreb’s at the beginning of each experiment. Agonists or antagonists were then added to the appropriate apical or basal domains. At the end of the experiments, bathing fluid and cells were removed and frozen at −20°C prior to cAMP measurements. cAMP was then quantitated by protein binding assay as previously described by Kapas and colleagues,20 and expressed as pmol/cm2 of cell monolayer.
Data analysis
All data are expressed as arithmetic mean (SEM) where n = number of monolayers. Statistical comparisons used the two tailed Student’s t test with p<0.05 being taken to represent a significant difference.
Drugs and materials
CT was obtained from the Swiss Serum and Vaccine Institute (Berne, Switzerland). 125I and 86Rb were obtained from Amersham Pharmacia Biotech (UK). ACh chloride, barium chloride, cAMP, dimethyl sulphoxide, forskolin, histamine dihydrochloride, hyoscine hydrobromide, IBMX, charybdotoxin, apamin, and vasoactive intestinal peptide were all obtained from Sigma (UK).
Immunohistochemistry
Materials
Formalin fixed paraffin embedded 4 μm transverse sections of ileum, mounted on chrome-alum coated glass slides, were collected from pathology archives. Tissue had been removed from five patients (median age 33 years (range 29–46)) at the time of right hemicolectomy for non-obstructing caecal tumours. A senior histopathologist had reported all ileal specimens as both macro and microscopically normal. Ethics permission for the use of archival material was obtained from the East London Health Authority ethics committee (T/02/027).
Methods
Primary antibodies to protein gene product 9.5 (PGP), a marker of total neural tissue, colocalised with choline acetyltransferase (ChAT), were used to identify cholinergic nerves in the mucosa and submucosa. Anti-M3 muscarinic receptor antibodies were used to localise M3 receptors in the same layers, using serial sections. Slides were deparaffinised using standard techniques. Antigen retrieval was achieved by incubation in 2 μg/ml proteinase K (Promega, Madison, Wisconsin, USA) at pH 7.6 and room temperature for 20 minutes. Sections were then rinsed in Tris buffered saline (TBS) at pH 7.6 and room temperature for five minutes before blocking non-specific binding of the first secondary antibody with 5% horse serum. Anti-PGP and anti-ChAT antibodies were then applied sequentially. Sections were initially incubated with primary anti-PGP antibodies (table 1 ▶) at 37°C for 90 minutes. Slides were washed in TBS and incubated with a secondary biotinylated antibody (Vectastain Universal Elite ABC kit, Vector laboratories, Peterborough, UK) at room temperature for 30 minutes and rinsed again in TBS.
Table 1.
Primary antibodies used for immunohistochemistry
| Antibody | Manufacturer | Type | Code | Raised in | Concn | Raised against | Antigen retrieval method |
| PGP 9.5 | Ultraclone UK | Monoclonal | 31A3 | Mouse | 1:400 | Human | Proteinase K |
| ChAT | Schemann, Hannover | Polyclonal | P3/YEB | Rabbit | 1:500 | Human | Proteinase K |
| M3 | Santa Cruz Biotechnology | Polyclonal | SC-7474 | Goat | 1:100 | Human | Microwave |
PGP 9.5, protein gene product 9.5; ChAT, choline acetyltransferase; M3, muscarinic type 3 receptor.
A tertiary peroxidase labelled avidin was applied at room temperature for 20 minutes, before developing with a Tyramide Signal Amplification Fluorescein system (NEN Life Science Products, Boston, Massachusetts, USA) for five minutes. Slides were washed with TBS and non-specific binding of the second secondary antibody was blocked using 5% goat serum. Sections were then incubated with primary anti-ChAT antibodies at 4°C for 18 hours and rinsed in TBS. A TRITC-immuno conjugated secondary antibody (Nordic Immunology, the Netherlands) was added for 30 minutes. Slides were finally mounted in Immuno-Mount (Shandon, Pittsburgh, Pennsylvania, USA).
M3 receptors
M3 receptor epitopes were exposed on serial sections, using microwave for four minutes in citrate buffer (pH 6.6) followed by localisation with anti-M3 antibodies using the method described previously for PGP.
Analysis
Sections were viewed using a Leica epifluorescence microscope (Leica imaging systems Ltd, Cambridge, UK) and appropriate filter blocks. Multiple digital image captures were taken of the mucosa, muscularis mucosa, and submucosa at ×200 magnification, using Leica DC200 software, in order to assess the distribution of staining and the degree of colocalisation of ChAT with PGP.
RESULTS
Response of T84 monolayers to CT and ACh
Transepithelial resistance (TER) of T84 monolayers was 595 (45) Ω/cm2. Basal Isc was 2 (1) μA/cm2. CT (0.0001–0.1 μg/ml) applied to the apical domain of T84 cell monolayers produced a concentration dependent increase in Isc (fig 1 ▶). Time to onset of effect and to peak effect were also dependent on concentration. When given basolaterally, the response to CT (0.1 μg/ml) reached a peak effect more rapidly (54 (4) μA/cm2 at 90 minutes; n = 5) compared with when given apically (46 (3) μA/cm2 at 130 minutes; n = 5, p<0.05). Compared with an aqueous control, CT decreased TER. For apically applied toxin, TER decreased from 406 (96) Ω/cm2 to 268 (32) Ω/cm2 (p<0.05, n = 5), and for basolaterally applied toxin, TER decreased from 633 (111) Ω/cm2 to 348 (48) Ω/cm2 (p<0.05, n = 5). Control values were 566 (41) Ω/cm2 to 513 (43) Ω/cm2 (p>0.05, n = 5). ACh (0.1–100 μM) applied basolaterally increased Isc from 1 (0) μA/cm2 (n = 4) to a maximum of 11 (3) μA/cm2 at 10 μM (n = 6). The responses were sided as apical application of ACh (10 μM) was ineffective (0 (0) μA/cm2, n = 4).
Figure 1.

Short circuit current (Isc) responses of T84 cells to cholera toxin (CT 0.0001 to 0.1 μg/ml) over 180 minutes. CT was applied to the apical surface unless otherwise indicated, when it was applied to the basolateral surface (B). Isc is expressed as change in current/cm2 of monolayer. Data are means (SEM) (n = 5).
Interaction of secretagogues
CT and forskolin both potentiated responses to ACh. In the presence of CT, responses of T84 cells to ACh were greatly potentiated. Figure 2 ▶ shows concentration response curves to ACh after pre-exposure of T84 cells to CT (0.01 μg/ml) or H2O vehicle (100 μl) for 180 minutes. Potentiation of ACh was dependent on the concentration of toxin. Responses to ACh (10 μM) ranged from 104 (9) μA/cm2 (n = 6) to 213 (11) μA/cm2 (n = 6), in the presence of 0.0001 and 0.1 μg/ml CT, respectively. Potentiation of ACh was also dependent on the exposure time of cell monolayers to CT. Responses to ACh (10 μM) varied from 26 (9) μA/cm2 (n = 6) to 43 (4) μA/cm2 (n = 6) after 30 minutes of exposure to CT (0.0001 μg/ml and 0.1 μg/ml, respectively). Hyoscine (1 μM given 30 minutes before ACh) abolished responses to ACh (10 μM; n = 6) given 180 minutes after CT (0.01 μg/ml). By itself, hyoscine had no effect on basal Isc or TER. Responses to ACh were also potentiated by forskolin. After five minutes of preincubation with forskolin (10 μM), responses to ACh (50 μM) were 111 (9) μA/cm2 (n = 6, p<0.05). Responses to ACh (50 μM) alone were 10 (1) μA/cm2 (n = 6).
Figure 2.

Short circuit current (Isc) responses of T84 monolayers to acetylcholine (ACh 0.1 μM–100 μM) given 180 minutes after pre-exposure to cholera toxin (CT 0.01 μg/ml) or vehicle. Responses to ACh in the presence of CT do not include the underlying response to the toxin. Data are means (SEM) (n = 4–6).
ACh potentiated forskolin but not CT. In the presence of ACh (10 μM) given 90 seconds before the toxin, the response to CT (0.01 μg/ml) after 180 minutes of exposure to the toxin was 27 (2) μA/cm2 (n = 6, p>0.05). In contrast, responses to forskolin (1.25 μM) after five minutes increased from 32 (2) μA/cm2 (n = 9) to 108 (11) μA/cm2 (n = 5, p<0.05) when ACh was given 90 seconds before forskolin.
CT also potentiated responses to histamine but not to VIP. In the presence of CT (0.01 μg/ml, 120 minutes of preincubation), responses to histamine (100 μM) increased from 14 (4) μA/cm2 (n = 6) to 60 (8) μA/cm2 (n = 6, p<0.05) while responses to VIP (0.3 nM) decreased from 51 (4) μA/cm2 to 28 (4) μA/cm2 (p<0.05, n = 6).
Effect of potassium channel block on secretagogue responses
Barium (5 mM) reduced responses to CT and forskolin but not to ACh (table 2 ▶). When barium was given 20 minutes before CT, subsequent responses obtained with ACh were unchanged (table 2 ▶). A combination of the highly specific calcium dependent potassium channel blockers charybdotoxin (0.5 μM) and apamin (0.5 μM) reduced responses to ACh in the presence of forskolin (table 2 ▶). None of the blockers used had any effect on basal Isc or TER.
Table 2.
Effect of the potassium channel blockers barium and charybdotoxin plus apamin on short circuit current (Isc) responses to cholera toxin, forskolin, and acetylcholine in T84 cells
| Control Isc response (μA/cm2) | Isc response in the presence of barium (μA/cm2) | Isc response in the presence of charybdotoxin+apamin (μA/cm2) | |
| CT 0.01 μg/ml (n = 5) | 34 (2) | 12 (1)* | ND |
| ACh 10 μM (n = 5) | 10 (3) | 12 (1) | ND |
| ACh 10 μM (180 min after CT 0.01 μg/ml; n = 5) | 133 (8) | 137 (26) | ND |
| Forskolin 10 μM (n = 6) | 22 (1) | 11 (1)* | 27 (2) |
| ACh 50 μM (5 min after forskolin 10 μM; n = 6) | 112 (9) | ND | 41 (3)* |
Barium (5 mM) and charybdotoxin (0.5 μM) plus apamin (0.5 μM) were given 20 minutes before secretogogues to the basolateral surface. Acetylcholine (ACh) was given 180 minutes after cholera toxin (CT) and five minutes after forskolin.
Data are means (SEM) (n = 6).
*p<0.05 compared with controls.
ND, no data.
Effect of secretagogues on basolateral 86Rb efflux and apical 125I efflux
Forskolin (10 μM) applied to the basolateral surface produced a rapid increase in the rate of 125I efflux which was significantly different from time matched controls. This coincided in time with the initial increase in Isc produced by forskolin. There was no significant change in efflux rate constants with ACh alone (50 μM). ACh (50 μM) applied to the basolateral surface, in the presence of forskolin (10 μM), produced a second increase in 125I efflux. This again was significantly different from time matched controls (fig 3 ▶, table 3 ▶).
Figure 3.

Effect of forskolin (B), acetylcholine alone (C), and acetylcholine in the presence of forskolin (D) on efflux of 125I from T84 cells. Acetylcholine (50 μM) was given five minutes after forskolin (10 μM) to the basolateral surface. The Y axis values are efflux rate constants (r/min). *p<0.05 compared with time matched controls (A). Data are means (SEM) (n = 6).
Table 3.
Effect of forskolin, acetylcholine, and acetylcholine in the presence of forskolin on efflux of 125I from T84 cells
| Treatment | Basal efflux rate | Peak efflux rate |
| None (n = 9) | 0.115 (0.076) | 0.092 (0.024) |
| Forskolin (n = 9) | 0.057 (0.014) | 0.231 (0.057)* |
| ACh (n = 7) | 0.102 (0.018) | 0.063 (0.011) |
| ACh in the presence of forskolin (n = 9) | 0.069 (0.012) | 0.166 (0.033)* |
Acetylcholine (ACh 50 μM) was given five minutes after forskolin (10 μM) to the basolateral surface. All values are apparent efflux rate constants (r/min), as described in the methods.
Data are means (SEM) (n = 9).
*p<0.05 compared with time matched basal controls.
Forskolin (10 μM) alone caused a small delayed but significant increase in 86Rb basolateral efflux compared with time matched controls. This was reduced in the presence of the potassium channel blocker barium. ACh also produced an increase in 86Rb efflux. In the presence of forskolin, ACh produced an additional increase which was significantly greater than that induced by ACh alone (fig 4 ▶, table 4 ▶). This was reduced by a combination of the calcium activated potassium channel blockers charybdotoxin plus apamin (table 4 ▶).
Figure 4.

Effect of forskolin (B), acetylcholine alone (C), and acetylcholine in the presence of forskolin (D) on efflux of 86Rb from T84 cells. Acetylcholine (50 μM) was given five minutes after forskolin (10 μM) to the basolateral surface. The Y axis values are efflux rate constants (r/min). *p<0.05 compared with time matched controls (A). †p<0.05 compared with peak 86Rb efflux induced by acetylcholine alone. Data are means (SEM) (n = 6).
Table 4.
Effect of the potassium channel inhibitors barium and charybdotoxin plus apamin on forskolin and acetylcholine induced efflux of 86Rb from T84 cells
| Treatment | Basal efflux rate | Peak efflux rate | Peak efflux in the presence of barium | Peak efflux in the presence of charybdotoxin plus apamin |
| None (n = 6) | 0.104 (0.007) | 0.107 (0.002) | 0.099 (0.005) | 0.103 (0.004) |
| Forskolin (n = 6) | 0.089 (0.010) | 0.127 (0.006)* | 0.107 (0.005)† | 0.121 (0.003) |
| ACh (n = 6) | 0.100 (0.004) | 0.186 (0.014)* | ND | ND |
| ACh in the presence of forskolin (n = 6) | 0.110 (0.008) | 0.242 (0.018)*‡ | ND | 0.170 (0.012)† |
Acetylcholine (ACh 50 μM) was given five minutes after forskolin (10 μM) to the basolateral surface. Barium (5 mM) and charybdotoxin (0.5 μM) plus apamin (0.5 μM) were given 20 minutes before agonists. All values are apparent efflux rate constants (r/min), as described in the methods.
Data are means (SEM) (n = 6).
*p<0.05 compared with time matched basal controls. †p<0.05 compared with peak efflux rates in the absence of antagonists. ‡p<0.05 compared with peak efflux induced by ACh alone.
Effect of CT and forskolin on cAMP levels
CT (0.01 μg/ml) and forskolin (1.25 μM) caused a time dependent increase in cAMP production in T84 cell monolayers. The effect of forskolin was significant after 10 minutes whereas CT took 120 minutes to reach significance (fig 5 ▶). Barium (5 mM) given 20 minutes before CT (0.01 μg/ml) had no effect on cAMP production by the toxin (fig 6 ▶) despite a considerable reduction in the Isc response (table 2 ▶). Barium alone has previously been shown to have no effect on basal cAMP levels.21 cAMP production was also unaffected by the presence of ACh (10 μM) given after 180 minutes pre-exposure to the toxin; ACh (10 μM) by itself had no effect on cAMP levels (fig 6 ▶).
Figure 5.

Measurement of cAMP production in T84 cells. Time course of the effect of cholera toxin (0.01 μg/ml) and forskolin (1.25 μM) on cAMP production was measured over 180 minutes and 60 minutes, respectively. cAMP levels are expressed as total nucleotide/cm2 of monolayer. Data are means (SEM) (n = 6). *p<0.05 compared with controls.
Figure 6.

Stimulation of cAMP production in T84 cells. cAMP levels (pmol/cm2 of monolayer) were measured at 180 minutes after exposure to cholera toxin (CT) or 3-isobutyl-1-methylxanthine (IBMX, controls), and at 30 minutes after exposure to acetylcholine (ACh). When given in combination, ACh was added to CT after 150 minutes and cAMP levels were measured at 180 minutes. Barium (5 mM) was given 20 minutes before CT (0.01 μg/ml). Data are means (SEM) (n = 6).
Distribution of cholinergic nerves and M3 receptors in human ileal mucosa
A high proportion of nerve fibres stained positively for ChAT (fig 7A ▶, 7B ▶) in all layers of the captured fields. The highest density of nerves within the mucosa was located towards the luminal aspect of the lamina propria associated, on serial sections, with M3 receptor immunoreactivity adjacent to the basolateral domain of enterocytes (fig 7C ▶). A high density of both ChAT positive nerves and M3 receptors were found in the muscularis mucosa. Nerve fibres and cell bodies stained positively for both PGP and ChAT in the submucosal plexus, with M3 receptors localised to submucosal blood vessels.
Figure 7.

Co-localisation (arrow) of protein gene product (PGP) (A) and choline acetyltransferase (ChAT) (B) in nerve fibres within the mucosa (m), muscularis mucosa (mm), and submucosa (sm) of normal ileum, with a high proportion of fibres staining positively for ChAT. The majority of fibres within the mucosa were located in the lamina propria, adjacent to the basolateral domain of enterocytes. Muscarinic M3 receptors (C) were localised, in serial sections, to the basolateral domain of enterocytes (arrow), muscularis mucosa, and blood vessels (arrowhead) of the submucosa. Bars represent 200 μm.
DISCUSSION
Acute infectious diarrhoea constitutes a worldwide health problem and continues to cause significant morbidity and mortality. The volume of diarrhoea produced in cholera is greater than most other types of diarrhoea and this is consistent with the proposal that a synergistic mechanism may play a role in the pathogenesis of the acute severe watery diarrhoea produced by Vibrio cholerae. Animal studies have demonstrated cholinergic nerves projecting to the mucosa.7,12,13 Cholinergic neurones have also been demonstrated around the crypts and extending up the villi of the human small intestine.22,23 However, both of these investigations used cholinesterase and acetylcholinesterase rather than choline acetyltransferase (ChAT) as markers for cholinergic neurones. Cholinesterase is not considered as reliable a marker for cholinergic neurones as ChAT as it can be associated with non-cholinergic neurones.24 In addition, functional studies support a secretory role for ACh in human and animal tissue.25–27 Using immunohistochemical staining of normal human ileum resection specimens, we have shown ChAT containing neurones and M3 muscarinic receptors within the submucosa and adjacent to the basolateral domain of epithelial cells. Such findings have not been described previously in the human small intestine. It is probable that CT releases ACh directly onto epithelial cells given the convincing evidence that the toxin activates a secretomotor neural reflex,4,5,28 and that stimulation of nerve fibres in human ileal mucosa cause atropine sensitive secretory responses.25
T84 colonic epithelial cells show a high degree of sensitivity to CT. Time to onset of Isc response as well as peak effect were concentration dependent. Increases in Isc to basolateral application of the toxin were unexpected as receptors for CT were originally shown to be located on the apical membrane of enterocytes.29 As basolaterally applied toxin achieved a peak effect more rapidly than apically applied toxin, it was considered unlikely that the toxin was crossing the monolayer. A more likely explanation would be that the CT receptor, a monosialoganglioside, is not restricted to the apical domain of enterocytes, which is not surprising for such a ubiquitous cell component.30 The earlier peak effect of basolaterally applied CT may be due to the location of adenylate cyclase on the basolateral domain of the enterocyte,31 in which case less time may be spent in intracellular trafficking of the toxin subunit. Alternatively, there may be more toxin ganglioside receptors on the basolateral membrane.32
In contrast with investigations using native tissue preparations,33,34 CT decreased the TER of T84 monolayers. Although increased responses to ACh in the presence of CT were seen after 180 minutes, we cannot exclude the possibility of tissue damage, especially as no index of cellular integrity was utilised.
Isc responses of T84 cells to ACh were dramatically increased in the presence of CT. Such potentiation was dependent on both the concentration and time of exposure of cells to the toxin. The Isc response to CT alone was shown to be associated with a time dependent increase in cAMP levels, an observation which is supported by earlier work where the authors also demonstrated a close correlation between Isc response and cAMP levels.2 It is likely that the magnitude of potentiation is dependent on cAMP levels and the results with forskolin would support such a conclusion. Higher concentrations of ACh were used with forskolin as these data were taken from radioisotopic efflux experiments where we wished to maximise the effects of secretagogues on ion efflux. The converse situation (that is, potentiation of cAMP dependent secretagogues by ACh) could only be demonstrated when using forskolin. This was probably due to the short lived effects of ACh disappearing by the time CT had initiated a significant increase in cAMP. Forskolin was shown to elevate cAMP levels within 10 minutes whereas a significant increase induced by CT was not shown until 120 minutes. The synergism described seems to be operating between secretagogues activating different second messenger pathways. Thus histamine, which in common with ACh is a calcium dependent secretagogue,35 was potentiated by prostaglandin E1 and Escherichia coli heat stable toxin, while VIP was not potentiated by CT. The interaction between CT and forskolin was also not additive.3
Understanding the nature of the synergistic interaction is complicated by the uncertainty regarding the mechanism of action of calcium dependent secretagogues. It is generally agreed that such compounds open basolateral potassium channels which hyperpolarise the cell and increase the electrical driving force for apical chloride secretion.36 However, there is controversy as to whether they open apical chloride channels. For instance, in cystic fibrosis intestine there is a failure of both cAMP and calcium dependent secretagogues to elicit chloride secretion yet this disease only affects the cAMP activated chloride channel.37 Similarly, Reenstra38 could not demonstrate carbachol induced chloride conductance in apical membranes of T84 cell monolayers. It was therefore proposed that the chloride conductance initiated by calcium dependent secretagogues was through constitutively open cAMP activated chloride channels.37 In contrast with these findings, Fuller and Benos have isolated and cloned a membrane protein from tracheal epithelium which is associated with a novel calcium activated chloride conductance when heterologously expressed.39 A porcine homologue has since been cloned and is expressed in porcine enterocytes.40 Moreover, Barrett and colleagues41 have produced evidence for an apical calcium activated chloride conductance which is both positively and negatively regulated by calcium dependent agonists. The negative regulation (that is, inhibition) is through generation of inositol tetrakisphosphate and would explain the short duration of action of ACh.
Potentiation of ACh by CT may occur at the level of the second messengers. For example, cAMP could inhibit the generation or actions of the negative regulator of calcium dependent secretion, inositol tetrakisphosphate,42,43 and thus relieve ACh of its autoinhibitory actions. However, the observation that even potentiated responses to ACh still remain very brief make such a mechanism unlikely. The cause of the synergism is more likely to result from optimum timing of ion channel opening.16 In the presence of CT the principal apical membrane chloride channel is open, thus allowing unhindered secretion of chloride ions when the driving force for such secretion is increased by opening of ACh activated basolateral potassium channels. This proposed mechanism of action has been supported in this study both by the characteristics of rubidium and iodine efflux during potentiation and also by the use of selective potassium channel inhibitors acting on calcium dependent potassium channels.
Forskolin rapidly increased the rate of apical 125I efflux from T84 cells. ACh alone did not have any effect on 125I efflux, in keeping with previous published data,38 although in the present investigation this may reflect the small responses in short circuit current which is an indication of chloride ion secretion. In the presence of forskolin, the Isc response to ACh was greatly potentiated and this coincided with a significant increase in 125I efflux. This suggests that the potentiation is associated with enhanced apical chloride secretion. Separately, both forskolin and ACh induce increases in basolateral 86Rb efflux. When given in the presence of forskolin, ACh induced 86Rb efflux was increased further, which supports the role of basolateral potassium channels in this mechanism. In T84 cells, the potassium channel blocker barium inhibits secretory responses to cAMP but not to calcium dependent secretagogues, by blocking separate and specific basolateral potassium channels.36,44 This was confirmed in the present investigation where barium attenuated responses to CT and forskolin, but not to ACh. This attenuated Isc response to forskolin was associated with a decrease in the rate of 86Rb efflux, with no effect on cAMP levels (using CT), confirming the role of basolateral potassium channels. Moreover, we demonstrated that barium had no effect on potentiation of ACh by CT despite attenuating short circuit responses to CT. To assess the role of calcium dependent potassium channels we investigated the effect of selective inhibition of such channels on the synergistic action of forskolin and ACh. A combination of charybdotoxin and apamin45 had no effect on forskolin Isc responses and 86Rb efflux but greatly attenuated the potentiated Isc responses and 86Rb efflux to ACh in the presence of forskolin. These results therefore suggest that the synergism between ACh and CT is dependent on cAMP activated apical chloride channels and calcium activated barium insensitive basolateral potassium channels. The cAMP dependent barium sensitive basolateral potassium channels are unlikely to play a significant role. By itself, barium does not appear to affect cAMP levels in enterocytes.21
In summary, the present investigation demonstrates a marked potentiation of the secretory actions of ACh by CT in human epithelial cells. Such an interaction may contribute to the overall secretory effects of the toxin as it has previously been shown to activate enteric secretomotor reflexes. In humans, these reflexes probably utilise ACh as a neurotransmitter at the neuroepithelial junction as we have shown that both cholinergic nerves and muscarinic M3 receptors are located at the basolateral domain of human ileal epithelial cells.
Acknowledgments
We are grateful to Professor LA Turnberg for useful discussions, Dr S Kapas for advice, facilities, and help with cAMP assays, and Ms Karin Fernandes for expert technical assistance with the cell culture. We also acknowledge Professor J Martin for supply of archived tissue samples. This project was supported by a grant from the Special Trustees of St Bartholomew’s Hospital and The Stanley Thomas Johnson Foundation.
Abbreviations
CT, cholera toxin
Isc, short circuit current
IBMX, 3-isobutyl-1-methylxanthine
VIP, vasoactive intestinal polypeptide
PGP, protein gene product
ChAT, choline acetyltransferase
TBS, Tris buffered saline
M3, muscarinic type 3 receptor
TER, transepithelial resistance
REFERENCES
- 1.Kimberg DV, Field M, Johnson J, et al. Stimulation of intestinal mucosal adenyl cyclase by cholera enterotoxin and prostaglandins. J Clin Invest 1971;50:1218–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett KE, Cohn JA, Huott PA, et al. Immune-related intestinal chloride secretion. II. Effect of adenosine on T84 cell line. Am J Physiol 1990;258:C902–12. [DOI] [PubMed] [Google Scholar]
- 3.Burleigh DE, Borman RA. Evidence for a nonneural electrogenic effect of cholera toxin on human isolated ileal mucosa. Dig Dis Sci 1997;42:1964–8. [DOI] [PubMed] [Google Scholar]
- 4.Cassuto J, Jodal M, Sjovall H, et al. Nervous control of intestinal secretion. Clin Res Rev 1981;1:11–21. [Google Scholar]
- 5.Jodal M, Holmgren S, Lundgren O, et al. Involvement of the myenteric plexus in the cholera toxin-induced net fluid secretion in the rat small intestine. Gastroenterology 1993;105:1286–93. [DOI] [PubMed] [Google Scholar]
- 6.Furness JB, Clerc N, Gola, et al. Identification of component neurons and organisation of enteric nerve circuits. In: Singer MV, Krammer HL, eds. Neurogastroenterology—from basics to the clinics. Dordrecht: Kluver Academic Publishers, 1999:134–47.
- 7.Clerc N, Furness JB, Li ZS, et al. Morphological and immunohistochemical identification of neurons and their targets in the guinea-pig duodenum. Neuroscience 1998;86:679–94. [DOI] [PubMed] [Google Scholar]
- 8.Dickinson KE, Frizzell RA, Sekar MC. Activation of T84 cell chloride channels by carbachol involves a phosphoinositide-coupled muscarinic M3 receptor. Eur J Pharmacol 1992;225:291–8. [DOI] [PubMed] [Google Scholar]
- 9.Przyborski SA, Levin RJ. Enterocytes on rat jejunal villi but not in the crypts posses m3 mRNA for the M3 muscarinic receptor localized by in situ hybridization. Exp Physiol 1993;78:109–12. [DOI] [PubMed] [Google Scholar]
- 10.O’Malley KE, Farrell CB, O’Boyle KM, et al. Cholinergic activation of Cl− secretion in rat colonic epithelia. Eur J Pharmacol 1995;275:83–9. [DOI] [PubMed] [Google Scholar]
- 11.Diener M, Knobloch SF, Bridges RJ, et al. Cholinergic-mediated secretion in the rat colon: neuronal and epithelial muscarinic responses. Eur J Pharmacol 1989;168:219–29. [DOI] [PubMed] [Google Scholar]
- 12.Li ZS, Furness JB. Immunohistochemical localisation of cholinergic markers in putative intrinsic primary afferent neurons of the guinea-pig small intestine. Cell Tissue Res 1998;294:35–43. [DOI] [PubMed] [Google Scholar]
- 13.Hens J, Schrodl F, Brehmer A, et al. Mucosal projections of enteric neurons in the porcine small intestine. J Comp Neurol 2000;421:429–36. [PubMed] [Google Scholar]
- 14.Porter AJ, Wattchow DA, Brookes SJ, et al. Choline acetyltransferase immunoreactivity in the human small and large intestine. Gastroenterology 1996;111:401–8. [DOI] [PubMed] [Google Scholar]
- 15.Ratcliffe EM, deSa DJ, Dixon MF, et al. Choline acetyltransferase (ChAT) immunoreactivity in paraffin sections of normal and diseased intestines. J Histochem Cytochem 1998;46:1223–31. [DOI] [PubMed] [Google Scholar]
- 16.Cartwright CA, McRoberts JA, Mandel KG, et al. Synergistic action of cyclic adenosine monophosphate- and calcium-mediated chloride secretion in a colonic epithelial cell line. J Clin Invest 1985;76:1837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Warhurst G, Higgs NB, Tonge A, et al. Stimulatory and inhibitory actions of carbachol on chloride secretory responses in human colonic cell line T84. Am J Physiol 1991;261:G220–8. [DOI] [PubMed] [Google Scholar]
- 18.Burleigh DE, Fernandes K, Perrett D. T(84) epithelial cells respond to 5-hydroxytryptamine when grown in serum-free media. Eur J Pharmacol 2000;390:103–6. [DOI] [PubMed] [Google Scholar]
- 19.Venglarik CJ, Bridges RJ, Frizzell RA. A simple assay for agonist-regulated Cl and K conductances in salt-secreting epithelial cells. Am J Physiol 1990;259:C358–64. [DOI] [PubMed] [Google Scholar]
- 20.Kapas S, Orford CD, Barker S, et al. Studies on the intracellular mechanism of action of α-melanocyte-stimulating hormone on rat adrenal zona glomerulosa. J Mol Endocrinol 1992;9:47–54. [DOI] [PubMed] [Google Scholar]
- 21.Hardcastle J, Hardcastle PT, Noble JM. The effect of barium chloride on intestinal secretion in the rat. J Physiol 1983;344:69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Issacs PET, Corbett CL, Riley AK, et al. In vitro behaviour of human intestinal mucosa - the influence of acetylcholine on ion transport. J Clin Invest 1976;58:535–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fang S, Wu R, Chrisrensen J. Intramucosal nerve cells in human small intestine. J Auton Nerv Syst 1993;44:129–36. [DOI] [PubMed] [Google Scholar]
- 24.Brown DR, Miller RJ. Neurohumoral control of fluid and electrolyte transport in intestinal mucosa. In: Schultz SG, ed. Handbook of physiology section 6—The gastrointestinal system, vol IV–Intestinal absorption. Bethesda: American Physiological Society, 1991:527–89.
- 25.Hubel KA, Shirazi S. Human ileal ion transport in vitro: changes with electrical field stimulation and tetrodotoxin. Gastroenterology 1982;83:63–8. [PubMed] [Google Scholar]
- 26.Chandan R, Megarry BH, O’Grady SM, et al. Muscarinic cholinergic regulation of electrogenic chloride secretion in porcine proximal jejunum. J Pharmacol Exp Ther 1991;257:908–17. [PubMed] [Google Scholar]
- 27.Cooke HJ. Influence of enteric cholinergic neurons on mucosal transport in guinea pig ileum. Am J Physiol 1984;246:G263–7. [DOI] [PubMed] [Google Scholar]
- 28.Cassuto J, Siewert A, Jodal M, et al. The involvement of intramural nerves in cholera toxin induced intestinal secretion. Acta Physiol Scand 1983;117:195–202. [DOI] [PubMed] [Google Scholar]
- 29.Critchley DR, Magnani JL, Fishman PH. Interaction of cholera toxin with rat intestinal brush border membranes. Relative roles of gangliosides and galactoproteins as toxin receptors. J Biol Chem 1981;256:8724–31. [PubMed] [Google Scholar]
- 30.Cuatrecasas P. Interaction of Vibrio cholerae enterotoxin with cell membranes. Biochemistry 1973;12:3547–58. [DOI] [PubMed] [Google Scholar]
- 31.Murer H, Ammann E, Biber J, et al. The surface membrane of the small intestinal epithelial cell. I. Localization of adenyl cyclase. Biochim Biophys Acta 1976;433:509–19. [DOI] [PubMed] [Google Scholar]
- 32.Fishman PH. Mechanism of action of cholera toxin: studies on the lag period. J Membr Biol 1980;54:61–72. [DOI] [PubMed] [Google Scholar]
- 33.Powell DW, Binder HJ, Curran PF. Active electrolyte secretion stimulated by choleragen in rabbit ileum in vitro. Am J Physiol 1973;225:781–7. [DOI] [PubMed] [Google Scholar]
- 34.Moriarty KJ, Higgs NB, Woodford M, et al. An investigation of the role of possible neural mechanisms in cholera toxin-induced secretion in rabbit ileal mucosa in vitro. Clin Sci (Lond) 1989;77:161–6. [DOI] [PubMed] [Google Scholar]
- 35.Wasserman SI, Barrett KE, Huott PA, et al. Immune-related intestinal Cl− secretion. I. Effect of histamine on the T84 cell line. Am J Physiol 1988;254:C53–62. [DOI] [PubMed] [Google Scholar]
- 36.Dharmsathaphorn K, Pandol SJ. Mechanism of chloride secretion induced by carbachol in a colonic epithelial cell line. J Clin Invest 1986;77:348–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anderson MP, Welsh MJ. Calcium and cAMP activate different chloride channels in the apical membrane of normal and cystic fibrosis epithelia. Proc Natl Acad Sci U S A 1991;88:6003–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reenstra WW. Inhibition of cAMP- and Ca-dependent Cl− secretion by phorbol esters: inhibition of basolateral K+ channels. Am J Physiol 1993;264:C161–8. [DOI] [PubMed] [Google Scholar]
- 39.Fuller CM, Benos DJ. Electrophysiological characteristics of the Ca2+-activated Cl− channel family of anion transport proteins. Clin Exp Pharmacol Physiol 2000;27:906–10. [DOI] [PubMed] [Google Scholar]
- 40.Gaspar KJ, Racette KJ, Gordon JR, et al. Cloning a chloride conductance mediator from the apical membrane of porcine ileal enterocytes. Physiol Genomics 2000;3:101–11. [DOI] [PubMed] [Google Scholar]
- 41.Barrett KE, Smitham J, Traynor-Kaplan A, et al. Inhibition of Ca(2+)-dependent Cl− secretion in T84 cells: membrane target(s) of inhibition is agonist specific. Am J Physiol 1998;274:C958–65. [DOI] [PubMed] [Google Scholar]
- 42.Xie W, Solomons KR, Freeman S, et al. Regulation of Ca2+-dependent Cl− conductance in a human colonic epithelial cell line (T84): cross-talk between Ins(3,4,5,6)P4 and protein phosphatases. J Physiol (Lond) 1998;510:661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie W, Kaetzel MA, Bruzik KS, et al. Inositol 3,4,5,6-tetrakisphosphate inhibits the calmodulin-dependent protein kinase II-activated chloride conductance in T84 colonic epithelial cells. J Biol Chem 1996;271:14092–7. [DOI] [PubMed] [Google Scholar]
- 44.Holliday ND, Cox HM. Modulation of chloride, potassium and bicarbonate transport by muscarinic receptors in a human adenocarcinoma cell line. Br J Pharmacol 1999;126:269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zygmunt PM, Hogestatt E.eds. Role of potassium channels in endothelium-dependent relaxation resistant to nitroarginine in the rat hepatic artery. Br J Pharmacol 1996;117:1600–6. [DOI] [PMC free article] [PubMed] [Google Scholar]