

Abstract
Background and aims: Interleukin-12 (IL-12), a p35/p40 heterodimer, plays a pivotal role in the immune response in Crohn’s disease (CD). Since IL-12 p40 dimers act as IL-12 antagonists, we assayed p40 dimer proteins to modulate chronic intestinal inflammation.
Methods: We generated a fusion protein consisting of the IL-12(p40) subunit fused to the constant region of IgG2b. IL-12(p40)-IgG2b was tested in a murine 2,4,6,-trinitrobenzene sulphonic acid (TNBS) colitis model and in lamina propria mononuclear cells (LPMNC) from patients with CD in vitro.
Results: Dimeric IL-12(p40)-IgG2b fusion protein bound specifically to the IL-12 receptor. In concentrations <10−7 M, it acted as an IL-12 antagonist as it inhibited interferon γ (IFN-γ) secretion, suppressed proliferation, and increased apoptosis of LPMNC from patients with CD. However, in concentrations >10−6 M, IL-12(p40)-IgG2b increased IFN-γ secretion and lymphocyte proliferation thereby acting as an IL-12 agonist. In TNBS colitic mice, IL-12(p40)-IgG2b decreased mortality (10% v 68%), prevented body weight loss, reduced tumour necrosis factor α, and increased IL-10 secretion.
Conclusions: The IL-12(p40)-IgG2b fusion protein has dichotomic properties as a specific IL-12 antagonist and selective repressor of mucosal inflammation at low concentration and as an IL-12 agonist at high concentration.
Keywords: interleukin 12, fusion protein, Crohn’s disease, inflammatory bowel disease
The pathophysiology of Crohn’s disease (CD) is characterised by an overriding cell mediated immune response which results in accumulation and activation of T lymphocytes and macrophages.1–2 By orchestrating the inflammatory process, proinflammatory cytokines such as interleukin (IL)-2, IL-12, and interferon γ (IFN-γ) are thought to promote the development of the disease.3–4
In active CD, expression of IL-12 receptor on mucosal cells is increased.5–6 IL-12, predominantly produced by antigen presenting cells,7 is secreted in mucosal tissues and contributes to the predominance of the Th1 response in patients with CD.8–10 Mucosal inflammation can be abrogated by administration of neutralising IL-12 antibodies in experimental models such as in 2,4,6,-trinitrobenzene sulphonic acid-(TNBS) induced colitis or in IL-2 deficient mice after immunisation with trinitrophenyl-KLH,11–12 indicating a key role of IL-12 in the development of the disease.
IL-12 is a heterodimeric protein composed of the p35 and p40 subunits. The IL-12(p40) subunit specifically inhibits IL-12 dependent cellular responses.13 The p40 homodimer, (p40)2, is 25–50-fold more active than the monomer in competing with native IL-12 in receptor binding in vitro14–15 and in vivo.16 Camoglio et al recently demonstrated that IL-12(p35) deficient mice develop a mild colitis compared with severe colitis in IL-12(p40) −/− mice on TNBS treatment, suggesting that IL-12(p40) dimers act contra-inflammatory.17 On the other hand, IL-12(p40)2 was reported to stimulate, rather than to inhibit, differentiation of CD8+ Th1 cells in vitro.18 Consequently, Th1 development is enhanced in allografted IL-12(p35) deficient mice.19–20
In order to modulate intestinal inflammation by blocking the IL-12 receptor, we generated a fusion protein that consisted of the IL-12 p40 subunit fused to the constant region of IgG2b. We demonstrated that the IL-12(p40)-IgG2b fusion protein binds to the IL-12 receptor, and in low concentrations inhibits inflammatory reactions of lamina propria mononuclear cells (LPMNC) from CD patients in vitro and represses experimental intestinal inflammation in vivo.
MATERIALS AND METHODS
Patients
Between July 1998 and July 2003, 32 patients treated at the University Hospital of the Saarland were included in our prospective study after written consent. Intestinal tissue specimens were obtained from two groups of patients: the control group included 15 specimens of macroscopically normal colonic mucosa from unaffected areas of colonic resections performed for adenocarcinoma; the second group included 17 affected colon specimens from patients with CD (see table 1 ▶).
Table1.
Patient characteristics
| Case No | Age (y) | Sex | Diagnosis | Duration (y) | Surgical treatment | Medical treatment |
| 1–15 | 53–72 | 8M/7F | Adenocarcinoma of ascending (n = 4) or descending (n = 11) colon | — | Hemicolectomy (right or left) | |
| 16 | 31 | M | CD, stricture in sigma | 7 | Resection of stricture | None |
| 17 | 25 | F | CD, ileocaecal conglomerate tumour | 1 | Resection of ileocaecal part | 5-ASA, glucocorticoids |
| 18 | 42 | M | CD, fistulas, colonic strictures | 12 | Right hemicolectomy | Glucocorticoids, azathioprine |
| 19 | 35 | M | CD, 2 strictures in terminal ileum | 5 | Resection of ileocaecal part | Glucocorticoids |
| 20 | 21 | F | CD, ileocaecal conglomerate tumour | 7 | Resection of ileocaecal part | 5-ASA |
| 21 | 23 | F | CD, ileocaecal conglomerate tumour | 5 | Resection of ileocaecal part | None |
| 22 | 35 | F | CD, ileocaecal conglomerate tumour | 4 | Resection of ileocaecal part | Glucocorticoids |
| 23 | 59 | M | CD, ileocaecal conglomerate tumour, jejunal stricture | 8 | Resection of ileocaecal part, resection of stricture | Glucocorticoids |
| 24 | 44 | M | CD, ileocaecal conglomerate tumour | 4 | Resection of ileocaecal part | Glucocorticoids |
| 25 | 43 | M | CD, ileocaecal conglomerate tumour | 7 | Resection of ileocaecal part | Glucocorticoids |
| 26 | 42 | M | CD, stricture in terminal ileum | 5 | Resection of ileocaecal part | 5-ASA, glucocorticoids |
| 27 | 34 | M | CD, ileocaecal conglomerate tumour | 3 | Resection of ileocaecal part | Glucocorticoids |
| 28 | 31 | M | CD, ileocaecal conglomerate tumour | 1 | Resection of ileocaecal part | Glucocorticoids |
| 29 | 43 | M | CD, conglomerate tumour | 12 | Resection of anastomosis | Glucocorticoids, azathioprine |
| 30 | 27 | F | CD, stricture in terminal ileum | 12 | Resection of stricture | Glucocorticoids, azathioprine |
| 31 | 30 | M | CD, conglomerate tumour | 9 | Resection of colonic anastomosis | Glucocorticoids, azathioprine |
| 32 | 21 | M | CD, stricture in terminal ileum | 4 | Resection of ileocaecal part | 5-ASA |
5-ASA 5-aminosalicylic acid, CD Crohn’s disease, F female, M male.
Cells, cell lines, and antibodies
The hybridoma OKT3 produces the anti-CD3 monoclonal antibody (mAb) (American Type Culture Collection, ATCC CRL 8001; Manassas, Virginia, USA) and the hybridoma 15E8 produces the anti-CD28 mAb (kindly provided by T van Lier, NCB, Amsterdam, the Netherlands). The anti-IL-12 receptor β1 and β2 chain mAbs were kindly provided by D Presky (Hoffmann-La Roche, Nutley, New Jersey, USA). Anti-CD4 (clone MT310) and anti-CD8 (clone DK25) antibodies were purchased from Dako (Hamburg, Germany). Recombinant human IL-2 and IL-12 were purchased from Pharmingen (Heidelberg, Germany). Peripheral blood mononuclear cells (PBMNC) and LPMNC were isolated from the large bowel, as described previously.21 Immunomagnetic beads (Dynal, Oslo, Norway) were used to isolate CD4+ or CD8+ peripheral blood lymphocytes (PBL) or LPMNC. Cell preparations with a purity of >95% were used.
Generation of the IL-12(p40)-IgG2b fusion protein
DNA coding for the IL-12(p40)-IgG2b fusion protein was generated by ligation of the mouse IL-12 p40 cDNA to the cDNA coding for the Fc part of human IgG2b. Briefly, mouse IL-12(p40) cDNA was amplified by polymerase chain reaction (PCR) using pSFG-mIL-12.p40.linker-delta.p35 DNA,22 kindly provided by Dr RC Mulligan (Whitehead Institute, Cambridge, Massachusetts, USA), as template and the primer oligonucleotides introducing SpeI and BamHI sites, respectively (table 2 ▶). The IL-12(p40) PCR product was digested with BamHI and SpeI and inserted into the XbaI/BamHI digested CD16-IgG2b-CDM8 vector DNA,23 containing the IgG2b cDNA fused to IL-2 cDNA, thereby replacing IL-2 cDNA by IL-12(p40) cDNA. To obtain the truncated IL-12(p40*)-IgG2b fusion protein with a deleted receptor binding site, IL-12(p40) cDNA was amplified by PCR techniques utilising an oligonucleotide primer 30 bases downstream of the translation start (table 2 ▶).
Table 2.
Primer oligonucleotides
| IL-12(p40)-IgG | |
| Upstream primer | agc ctg act agt atg tgg gag ctg gag aaa gac |
| Downstream primer | atg ctc tgg atc cga tcg gac cct gca ggg aac |
| Truncated IL-12(p40*)-IgG | |
| Upstream primer | agc ctg act agt acc atc act gtc aaa gag |
| Downstream primer | atg ctc tgg atcc ga tcg gac cct gca ggg aac |
For protein expression, 293 cells (2×106 cells) were transfected with 2 µg of plasmid DNA and the culture supernatant was harvested at day 7. Fusion protein from the supernatant was bound to protein A-Sepharose CL4B (0.5 g/l) (Pharmacia Biotech, Piscataway, New Jersey, USA) for 16 hours at 4°C, pH 7.4, washed with 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, and eluted with 1 M acetic acid. Purified proteins were free of detectable amounts of endotoxins (Kinetic-Chromogenic Lysate, BioWhittaker Inc, Walkersville, Maryland, USA).
Detection of the IL-12(p40)-IgG2b fusion protein
The IL-12(p40)-IgG2b protein was detected by ELISA. Briefly, 96 well microtitre plates were coated with rat antimouse Fc antiserum (5 µg/ml phosphate buffered saline (PBS); Dako, Hamburg, Germany), blocked with 3% (w/v) bovine serum albumin in PBS, and incubated with serial dilutions of cell supernatant containing fusion protein or human IgG (0.5 µg/ml) as a control. Plates were incubated with a rat antihuman Fc polyclonal antibody conjugated with alkaline phosphatase (1 µg/ml PBS; Dako). ELISA was developed by addition of the alkaline phosphatase substrate disodium p-nitro-phenyl phosphate (1 mg/ml) (Sigma, Deisenhofen, Germany) in 0.9 M diethanolamine, 0.5 mM MgCl2, pH 9.8.
For Western blot analysis, 5 µg of purified fusion protein was electrophoresed through a 10% sodium dodecyl sulphate-polyacrylamide gel and electrophoretically transferred onto nitrocellulose membranes. Membranes were blocked with Tris buffered saline, 5% (w/v) bovine serum albumin, incubated with an anti-IL-12p40/p70 antibody (clone C8.6; 1:250) (Pharmingen), and subsequently with a goat antimouse Ig antibody conjugated to horseradish peroxidase (1:5000) (Dianova, Hamburg, Germany).
Binding of the IL-12(p40)-IgG2b fusion protein to IL-12 receptor positive cells
PBMC were stimulated for 72 hours with human IL-2 (400 U/ml) and phorbol myristate acetate (PMA) (2 ng/ml). Cells were washed, incubated with fetal calf serum (10% v/v) for 15 minutes at 4°C to block non-specific staining, and incubated with fusion protein in increasing concentrations. Bound fusion protein was monitored by an FITC conjugated anti-IgG antibody (Dako). Specific binding of the fusion protein was confirmed by competitive inhibition with recombinant IL-12 (Pharmingen). Cells were stained for CD4, CD8, and IL-12 receptor β1 and β2 chains, respectively, fixed for 30 minutes with 1% (v/v) paraformaldehyde, and analysed by three colour flow cytometry (FACSVantage; Becton Dickinson, Heidelberg, Germany) using CellQuest software.
Proliferation assays
Cells (1×105) in 0.2 ml RPMI 1640 medium and 10% fetal calf serum were incubated with 0.5 µCi [3H]thymidine (specific activity 3.64 GBq/mg) for 16 hours at 37°C. Cells were harvested on a cell harvester and [3H]thymidine incorporation was determined by liquid scintillation spectrometry. All assays were performed in triplicate with an intra-test variance of less than 15%.
Determination of IFN-γ secretion and staining of intracellular cytokines
IFN-γ in culture supernatant was determined by ELISA according to the manufacturer’s instructions (BD Pharmingen). Measurement of intracellular cytokines was performed as described previously.27
Detection of caspase-3 positive cells and apoptosis
Apoptotic cells were detected by flow cytometry as described previously.24 Briefly, cells were washed twice in PBS, incubated in 0.1% (w/v) sodium citrate, 0.1% (v/v) Triton X-100, and propidium iodide (50 µg/ml) for 4–8 hours at 4°C, and analysed by flow cytometry. Apoptotic cells were determined by monitoring cells with hypodiploid nuclei. Active caspase-3 was assayed by FACS analysis. Briefly, cells were fixed, permeabilised, and incubated with a rabbit antibody against amino acids 163–175 of murine caspase-3 (Biocarta Europe, Hamburg, Germany) and subsequently a FITC labelled goat antirabbit antibody.
Induction of TNBS colitis
Balb/c mice (female, six weeks old) were obtained from Charles River (Sulzfeld, Germany). TNBS colitis was induced as described previously.11,25,26 Briefly, TNBS (2.0 mg) (Sigma) in 50% ethanol was slowly administered into the lumen of the colon via a 3.5 F catheter. Mice were treated by daily intraperitoneal injections of IL-12(p40)-IgG2b (20 µg/200 µl PBS and 2 µg/200 µl PBS, respectively). Treatment was started two hours after application of TNBS and continued until day 6. Mice in the control groups were injected with truncated IL-12(p40*)-IgG2b (20 µg/200 µl PBS) fusion protein.
Grading of histological changes
Cryostat sections (4 µm) of tissues were mounted on APS treated glass slides, air dried, fixed in 4% (wt/vol) paraformaldehyde/PBS (pH 7.4), and stained with haematoxylin-eosin. The degree of inflammation of colon specimens was graded semiquantitatively from 0 to 4, as described by Neurath and colleagues.11 Grading was performed in a blinded fashion by AS.
Statistical analysis
Statistical analyses were performed using the Wilcoxon rank test or χ2 test. Data were considered to be significant at the p⩽0.05 level.
RESULTS
IL-12(p40)-IgG2b fusion protein is a dimeric protein and binds specifically to IL-12 receptor positive cells
We generated the IL-12(p40)-IgG2b fusion protein by joining the p40 domain to the Fc domain of IgG2b, as described in the materials and methods section. For control purposes, we deleted the receptor binding site of IL-12(p40) and, by fusion to Fc IgG2b, we generated the truncated fusion protein IL-12(p40*)-IgG2b. Western blot analysis revealed that the IL-12(p40)-IgG2b fusion protein had an apparent molecular weight of 140 kDa under non-reducing conditions and of 70 kDa under reducing conditions (fig 1 ▶), indicating that the IL-12(p40)-IgG2b fusion protein forms homodimers, probably via disulphide bonds between the Fc domains.
Figure 1.
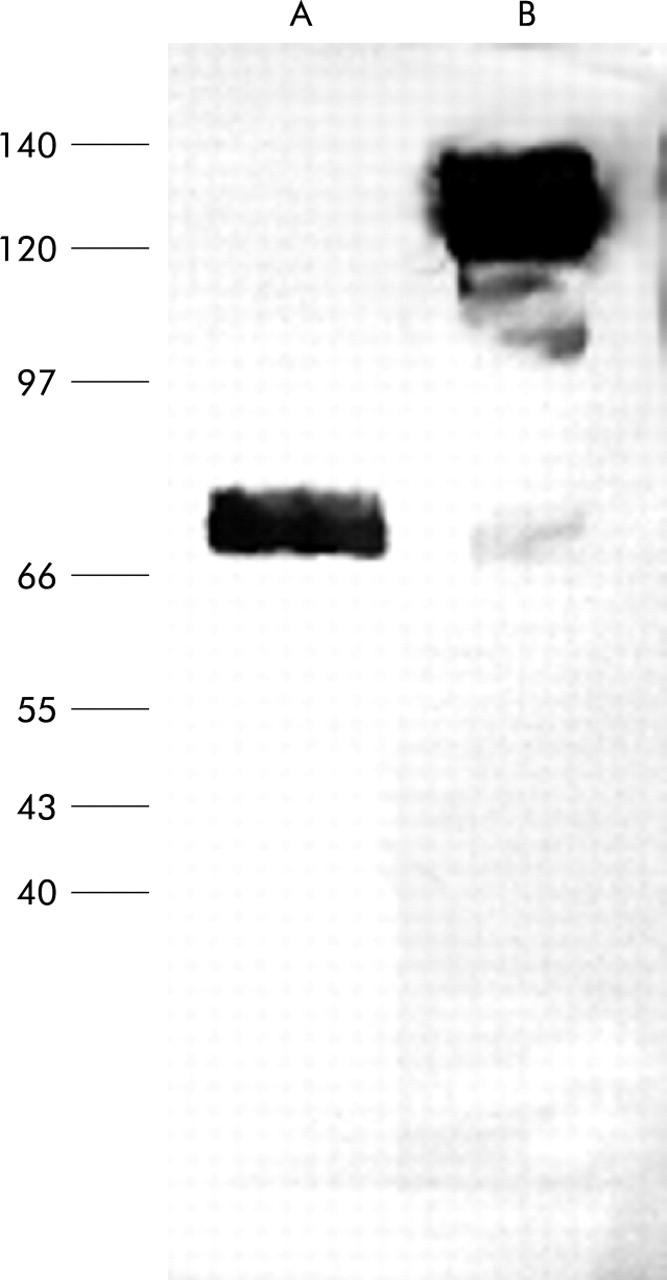
Western blot analysis of the recombinant IL-12(p40)-IgG2b fusion protein. The IL-12(p40)-IgG2b fusion protein was expressed in 293 cells, purified from the culture supernatant, and electrophoretically separated in a sodium dodecyl sulphate-polyacrylamide gel under reducing (A) and non-reducing (B) conditions. After western blotting, the fusion protein was detected by the anti-IL-12p40/p70 antibody C.8.6.
To test whether the IL-12(p40)-IgG2b fusion protein binds to IL-12 receptor positive cells, PBL were stimulated with IL-2 plus PMA for 72 hours in vitro to induce IL-12 receptor expression. FACS analyses using antibodies against IL-12 receptor β1 and β2 chains indicated that 47–83% (median 65%) of CD4+ T cells expressed both chains of the IL-12 receptor. IL-12(p40)-IgG2b fusion protein bound in a concentration dependent manner to activated CD4+ T cells whereas the truncated IL-12(p40*)-IgG2b protein did not (fig 2 ▶). Incubation with recombinant IL-12 competed with IL-12(p40)-IgG2b for binding, indicating IL-12 receptor specific binding of the fusion protein.
Figure 2.

IL-12(p40)-IgG2b fusion protein, but not the truncated IL-12(p40*)-IgG2b protein, binds to activated peripheral blood lymphocytes (PBL). PBL were activated with IL-2 (400 U/ml) plus phorbol myristate acetate (2 ng/ml) for 72 hours. Approximately 65% of cells expressed both chains of the IL-12 receptor (data not shown). Subsequently, PBL were incubated with increasing concentrations of IL-12(p40)-IgG2b and the truncated IL-12(p40*)-IgG2b fusion protein. Binding of the fusion protein was recorded by FACS analysis using an FITC conjugated antihuman IgG antibody. Median and minimum/maximum values of five independent experiments are indicated.
IL-12(p40)-IgG2b decreases IFN-γ secretion of LPMNC
We determined whether blocking of the IL-12 receptor by the IL-12(p40)-IgG2b fusion protein modulated IFN-γ secretion of activated LPMNC in vitro. Freshly isolated LPMNC cells were stimulated with immobilised anti-CD3 and anti-CD28 mAb plus increasing concentrations of IL-12 (20–2000 pg/ml) to secrete IFN-γ (fig 3 ▶). Addition of increasing amounts of IL-12(p40)-IgG2b fusion protein (2 pg/ml–200 ng/ml) resulted in a concentration dependent inhibition of IFN-γ secretion by activated LPMNC. In concentrations >10−6 M, however, IL-12(p40)-IgG2b increased IFN-γ secretion in LPMNC, indicating an agonistic effect of the fusion protein. The effect was mediated via the IL-12 receptor because addition of the truncated IL-12(p40*)-IgG2b fusion protein that does not bind to the IL-12 receptor did not modulate IFN-γ secretion of cells (fig 3 ▶).
Figure 3.

IL-12(p40)-IgG2b fusion protein (FP) inhibits IL-12 induced interferon γ (IFN-γ) secretion in activated lamina propria mononuclear cells (LPMNC). LPMNC from a control patient were activated by incubation with an anti-CD3 plus an anti-CD28 antibody for 72 hours, and recombinant IL-12 (2, 20, 200, and 2000 pg/ml) was added. IFN-γ concentrations in culture supernatants after 72 hours were determined by ELISA. Addition of IL-12(p40)-IgG2b fusion protein in increasing concentrations (2 pg/ml–20 μg/ml) decreased the amount of secreted IFN-γ. Interestingly, addition of IL-12(p40)-IgG2b at high concentrations (20 µg/ml) resulted in a substantial increase in IFN-γ secretion. A representative experiment of four independent experiments is shown (patients: n = 4).
To characterise the agonistic effect, LPMNC were separated in CD4+ and CD8+ T cell populations by negative purifications procedures and incubated with the IL-12(p40)-IgG2b fusion protein. IFN-γ was induced on incubation of only CD8+ T cells and not CD4+ T cells. Even high concentrations of IL-12(p40)-IgG2b did not induce IFN-γ secretion in CD4+ LPMNC. As a control, these cells secreted significant amounts of IFN-γ after stimulation with PMA plus ionomycin (data not shown).
IL-12(p40)-IgG2b decreases proliferation and increases apoptosis of LPMNC of patients with CD
In the next set of experiments we studied whether the fusion protein modulates proliferation of activated LPMNC in vitro. LPMNC from patients with CD were stimulated with immobilised anti-CD3 mAb plus anti-CD28 mAb. Addition of the IL-12(p40)-IgG2b protein (0.02 pg/ml–2 ng/ml) inhibited cell proliferation in a concentration dependent manner (fig 4 ▶). However, higher concentrations of the fusion protein increased proliferation of LPMNC. Addition of recombinant IL-12 (2 ng/ml) completely abolished the IL-12(p40)-IgG2b induced growth inhibition, indicating that the effect was specifically mediated via the IL-12 receptor.
Figure 4.

IL-12(p40)-IgG2b fusion protein (FP) inhibits proliferation of lamina propria mononuclear cells (LPMNC) from patients with Crohn’s disease (CD). LPMNC from a patient with CD were activated by incubation with an immobilised anti-CD3 plus a soluble anti-CD28 antibody. Recombinant IL-12 (20 pg/ml) was added for 72 hours in the presence of increasing concentrations of IL-12(p40)-IgG2b fusion protein. Cell proliferation was quantified by [3H]thymidine incorporation, as described in the material and methods section. The effect of the IL-12(p40)-IgG2b fusion protein was abolished by addition of recombinant IL-12 (2000 pg/ml). A representative experiment of three independent experiments is shown. Mean (SEM) values are indicated.
To obtain further insight into the regulation of fusion protein mediated growth arrest, we monitored the number of apoptotic LPMNC from patients with CD and from healthy controls. In controls (n = 5), activation of LPMNC with the anti-CD3 plus anti-CD28 antibody increased the number of capase-3 positive cells (+22% (median); range 5–48%), as monitored by FACS analysis. Addition of the fusion protein together with αCD3 plus αCD28 stimulation did not increase this rate significantly (median +43% (range 6–53%)). In patients with CD (n = 5), activation of LPMNC by stimulation with anti-CD3 plus anti-CD28 antibodies did not alter the number of capase-3 positive cells (−4% (−23–26%)). Activation in the presence of the fusion protein however increased caspase-3 positive cells significantly (+40.1% (13.0–70.0)). In contrast, incubation of LPMNC from both groups with the truncated fusion protein IL-12(p40*)-IgG2b did not exhibit any specific effect (data not shown).
Stimulation of LPMNC with anti-CD3 plus anti-CD28 antibodies resulted in a frequency of apoptotic cells of 33% (range 18–78%) (fig 5 ▶). Addition of IL-12(p40)2-IgG2b increased the rate of apoptotic cells significantly. IL-12(p40)-IgG2b increased apoptosis is best reflected in the mean per cent increase value (difference between IL-12(p40)-IgG2b αCD3 plus αCD28 stimulation induced apoptotic rate and αCD3 plus αCD28 stimulation induced apoptotic rate as a percentage of the activation induced apoptosis value) where IL-12(p40)-IgG2b resulted in an increase of 47% (range 10–124%) of the apoptotic rates (n = 10). In patients with active CD (n = 8), activation of LPMNC did not increase the percentage of apoptotic cells significantly (fig 5 ▶), as previously described by Boirivant and colleagues.24 However, addition of IL-12(p40)2-IgG2b accelerated apoptosis significantly (+39% (range 5–48%)).
Figure 5.

IL-12(p40)-IgG2b fusion protein accelerates apoptosis in lamina propria mononuclear cells (LPMNC) from patients with Crohn’s disease (CD). LPMNC from patients with CD (n = 8) and control patients (n = 10) were activated by incubation with an immobilised anti-CD3 monoclonal antibody (mAb) and anti-CD28 mAb for 24 hours. In addition, cells were incubated with IL-12(p40)-IgG2b fusion protein (0.2 µg/ml) and the number of cells that entered apoptosis was determined by flow cytometry. Median values are shown. Numbers above the bars indicate minimum and maximum values.
IL-12(p40)-IgG2b fusion protein abrogates TNBS induced colitis
In a series of in vivo studies we addressed the question of whether blocking IL-12 receptor by administration of IL-12(p40)-IgG2b fusion protein affects the clinical course of TNBS induced colitis. In a treatment trial, the IL-12(p40)-IgG2b fusion protein (20 µg and 2 µg, respectively, per mouse per day) was intraperitoneally injected into Balb/c mice starting two hours after induction of TNBS colitis. As summarised in fig 6 ▶, 68% (20.0 µg) and 63% (2.0 µg), respectively, of mice with TNBS colitis and administration of IL-12(p40)-IgG2b fusion protein survived to day 6 compared with 10% of TNBS colitis mice treated with the truncated IL-12(p40*)-IgG2b fusion protein (p⩽0.01). Moreover, TNBS colitis mice that received IL-12(p40)-IgG2b fusion protein regained their initial body weight at day 6 whereas TNBS colitis mice treated with the truncated fusion protein continuously lost their body weight (data not shown). In addition, IL-12(p40)-IgG2b treated mice were more active and lost their ruffled coat appearance associated with TNBS colitis compared with untreated mice and mice inoculated with the truncated fusion protein.
Figure 6.

IL-12(p40)-IgG2b fusion protein increases survival of Balb/c mice after induction of 2,4,6,-trinitrobenzene sulphonic acid (TNBS) colitis. TNBS colitis was induced in Balb/c mice as described above and IL-12(p40)-IgG2b fusion protein (20 µg and 2 µg, respectively, per mouse per day) was intraperitoneally injected everyday starting two hours after induction of TNBS colitis. As controls, the truncated IL-12(p40*)-IgG2b fusion protein was administered. Survival rate is shown in a Kaplan-Meier plot. The graph represents data from four independent experiments with at least six mice in each group. Application of IL-12(p40)-IgG2b fusion protein at both concentrations decreased the mortality rate of TNBS colitis mice significantly (p⩽0.01; χ2 test).
The pathological changes in mice after induction of TNBS colitis have been described in detail previously.11 In these animals, moderate colitis is characterised by an inflammatory infiltrate in the colonic lamina propria and increased mitotic activity in the elongated epithelial crypts. In severe disease, the cellular infiltrate is composed of both mono and polymorphonuclear cells and is present in all layers of the colonic gut wall. Crypt elongations and destructions are also seen (fig 7A ▶). After treatment with IL-12(p40)-IgG2b fusion protein (20 µg/day), TNBS induced colonic inflammation is abrogated and a nearly normal histological appearance of the colon is restored (fig 7B ▶). This observation was confirmed by histological grading of colon sections: data from three independent experiments showed significant reduction in inflammatory activity after treatment with the IL-12(p40)-IgG2b fusion protein—that is, the score of inflammation was 3.0 (1.0–4.0) in the TNBS group compared with 1.5 (0–3.0) in IL-12(p40)-IgG2b treated mice (p⩽0.05).
Figure 7.

Histological analysis of colonic specimens at day 7 after induction of 2,4,6,-trinitrobenzene sulphonic acid (TNBS) colitis. Histopathology of colon specimens of mice with TNBS induced colitis (A) and TNBS plus IL-12(p40)-IgG2b treated mice (B). A pronounced inflammatory infiltrate with elongation and destruction of crypts is obvious in mice after treatment with truncated fusion protein. Treatment with IL-12(p40)-IgG2b abrogated TNBS induced inflammation and restored a normal histological appearance of the colon (microscope magnification: B, 140×; A, 200×).
Administration of IL-12(p40)-IgG2b fusion protein decreases TNF-α and increases IL-10 secretion in mice with TNBS colitis
We investigated whether IL-12(p40)-IgG2b fusion protein modulates cytokine secretion of spleen T cells in mice with TNBS colitis. As summarised in fig 8 ▶, the number of tumour necrosis factor α (TNF-α) positive CD4+ T cells was decreased in colitis mice treated with IL-12(p40)-IgG2b fusion protein (20.0 µg per mice per day) compared with mice after treatment with the truncated IL-12(p40*)-IgG2b fusion protein (p⩽0.05). In addition, a much larger fraction of CD4+ T cells from TNBS colitis mice treated with IL-12(p40)-IgG2b fusion protein stained positive for IL-10 compared with colitis mice without treatment with fusion protein (p⩽0.05).
Figure 8.

IL-12(p40)-IgG2b fusion protein reduces the number of tumour necrosis factor α (TNF-α) and increases the number of IL-10 producing spleen T cells in 2,4,6,-trinitrobenzene sulphonic acid (TNBS) colitis mice. Mononuclear cells from the spleen were stimulated for four hours with phorbol myristate acetate (50 nM) and ionomycin (250 ng/ml) in the presence of monensin to block exocytosis. Mice were treated with IL-12(p40)-IgG2b fusion protein and the truncated IL-12(p40*)-IgG2b fusion protein as a control (each 20 µg/day/mouse) for five days. Spleen cells were stained for intracellular cytokines and recorded by FACS analysis. The number of cytokine expressing CD4+ T cells from mice without TNBS application (controls), from TNBS colitic mice after application of the truncated IL-12(p40*)-IgG2b fusion protein, and from colitic mice after application of IL-12(p40)-IgG2b fusion protein were determined. Mean (SD) values are shown. p<0.05 compared with TNBS colitic mice after application of the truncated IL-12(p40*)-IgG2b fusion protein. IFN-γ, interferon γ.
DISCUSSION
We generated the fusion protein IL-12(p40)-IgG2b that consists of the IL-12(p40) subunit fused to the IgG Fc domain which specifically binds to the IL-12 receptor and represses IFN-γ secretion and cell proliferation of prestimulated LPMNC in vitro. The IL-12 antagonistic activity of IL-12(p40)-IgG2b is dose dependent in the range 2 pg/ml–0.2 μg/ml (that is, 10−9–10−7 M). In concentrations ⩾10−6 M however the IL-12(p40)-IgG2b fusion protein exhibits IL-12 agonistic activities, indicated by induction of IFN-γ secretion. This is at least partially based on induction of STAT-4 phosphorylation (data not shown) resulting in transactivation of the IFN-γ promoter.27 The concentration dependent dichotomic properties of the fusion protein are specific because the IL-12(p40*)-IgG2b protein without the IL-12 receptor binding site does not exhibit these activities.
One of the most striking observations is that TNBS induced colitis in mice can be successfully prevented by the IL-12(p40)-IgG2b fusion protein. The survival rate at day 6 increased from 10% without treatment to more than 60% with IL-12(p40)-IgG2b treatment. This was accompanied by significant alterations in the number of cytokine secreting spleen cells that however may not be representative of LPMNC. In TNBS colitis mice treated with the IL-12(p40)-IgG2b fusion protein however the number of TNF-α secreting CD4+ T cells was similar compared with that in non-colitis mice—that is, 30% of CD4+ T cells compared with 50% of CD4+ T cells in colitis mice treated with the truncated IL-12(p40*)-IgG2b protein produced TNF-α. The decrease in TNFα positive cells was paralleled by a substantial increase in the number of IL-10 secreting cells. A number of experimental models indicate that IL-10 is involved in the prevention of T cell mediated inflammation in the gut.28–30 However, recent studies question the role of IL-10 as an anti-inflammatory therapeutic agent in human inflammatory bowel disease.31–34 Interestingly, IL-12(p40)-IgG2b treatment resulted in an increase in IFN-γ positive cells (see fig 8 ▶). Similar effects were observed in TNBS colitis after protective treatment with anti-CD44v7 antibodies.25 The IL-12(p40)-IgG2b fusion protein moreover increased substantially the number of apoptotic lamina propria cells from patients with CD, thereby limiting the inflammatory immune response. Although several studies indicate that lamina propria T cells are resistant to apoptosis,24,35,36 our results clearly demonstrated induction of caspase-3 positive cells and apoptosis by the IL-12(p40)-IgG2b fusion protein.
Alternative strategies for the treatment of CD include TNF-α neutralising antibodies.37,38 Clinical trials to date have been disappointing as only a minority of patients have undergone stable clinical remission39,40 and the rate of complications, such as lethal tuberculosis, increased.41,42 It is tempting to speculate that these complications may not be as frequent after treatment with IL-12(p40)-IgG2b fusion protein. Moreover, IL-12(p40) is protective particularly in mycobacterial infections.43,44 IL-12(p40) deficient mice are more susceptible to infection with Listeria monocytogensis than p35 deficient mice.19,20 Clearance of an intracellular bacterium, Francisella tularensis, is dependent on the IL-12 p35p40 heterodimer and the p40 dimer,45 the latter activate macrophages, but not CD4+ or CD8+ T cells or natural killer cells. This indicates that IL-12(p40) does not inhibit reactions of innate immunity.46 Taken together, administration of IL-12(p40)-IgG2b fusion protein is not expected to be associated with eruption of lethal bacterial infections.
Although IL-12 neutralisation by antibodies is highly effective in experimental models of Th1 dominated inflammation,7,47,48 blocking the IL-12 receptor with IL-12(p40)-IgG2b would offer several advantages: (i) syngeneic domains of the fusion protein will avoid immune reactions against the protein itself, and (ii) due to the 140 kDa dimer, an expected long serum half life time compared with certain cytokine fusion proteins.49
In summary, we have demonstrated that the engineered fusion protein IL-12(p40)-IgG2b profoundly alters the immune response in a murine Th1 model of intestinal inflammation and inhibits proinflammatory reactions of human lamina propria lymphocytes of patients with CD. As a cautionary note however high concentrations of the IL-12(p40)-IgG2b fusion protein act agonistic to IL-12 as they promote IFN-γ secretion and increase T cell proliferation. Further investigations on the molecular action of the IL-12(p40)-IgG2b fusion protein may provide us with a more robust agent to modulate chronic inflammatory bowel diseases with tolerable side effects.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsgemeinschaft (DFG Sta 295/4-1 and 295/5-1) and the Competence network “Inflammatory bowel disease”, Core facility Homburg “Animal models”. AS was a recipient of a Heisenberg professorship of the DFG (Sta 295/3-1). The Zentrum für Molekulare Medizin Köln (ZMMK), the Deutsche Forschungsgemeinschaft, and the Deutsche Krebshilfe supported work in the HA laboratory. The authors thank the medical staff of the Department of Surgery (Head: Professor Dr M Schilling), Saarland University, for providing surgical specimens. We are indebted to R Goebel and B Ludwig for technical assistance.
Abbreviations
5-ASA, 5-aminosalicylic acid
CD, Crohn’s disease
TNBS, 2,4,6,-trinitrobenzene sulphonic acid
LPMNC, lamina propria mononuclear cells
PBMNC, peripheral blood mononuclear cells
IL, interleukin
IFN-γ, interferon γ
mAb, monoclonal antibody
PCR, polymerase chain reaction
PBS, phosphate buffered saline
PMA, phorbol myristate acetate
PBL, peripheral blood lymphocytes
TNF-α, tumour necrosis factor α
REFERENCES
- 1.Zeitz M. Pathogenesis of inflammatory bowel disease. Digestion 1997;58(suppl 1):59–61. [DOI] [PubMed] [Google Scholar]
- 2.Fiocchi C. Inflammatory bowel disease: Etiology and pathogenesis. Gastroenterology 1998;115:182–205. [DOI] [PubMed] [Google Scholar]
- 3.Breese E, Braegger CP, Corrigan CJ, et al. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology 1993;78:127–31. [PMC free article] [PubMed] [Google Scholar]
- 4.Fuss IJ, Neurath M, Boirivant M, et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunology 1996;157:1261–70. [PubMed] [Google Scholar]
- 5.Parrello T, Montelleone G, Cucchiara S, et al. Up-regulation of the IL-12 receptor beta 2 chain in Crohn’s disease. J Immunoogy 2000;165:7234–9. [DOI] [PubMed] [Google Scholar]
- 6.Stallmach A, Marth T, Adrian T, et al. Increased expression of IL-12 receptor β2 mRNA in lamina propria mononuclear cells of patients with active Crohn’s disease. Int J Colorectal Dis 2002;17:303–10. [DOI] [PubMed] [Google Scholar]
- 7.Gately MK, Renzetti LM, Magram J, et al. The interleukin-12/interleukin-12-receptor system: Role in normal and pathologic immune responses. Annu Rev Immunol 1998;16:495–521. [DOI] [PubMed] [Google Scholar]
- 8.Monteleone G, Biancone L, Marasco R, et al. Interleukin 12 is expressed and actively released by Crohn’s disease intestinal lamina propria mononuclear cells. Gastroenterology 1997;112:1169–78. [DOI] [PubMed] [Google Scholar]
- 9.Parronchi P, Romagnani P, Annunziato F, et al. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol 1997;150:823–32. [PMC free article] [PubMed] [Google Scholar]
- 10.Berrebi D, Besnard M, Fromont-Hankard G, et al. Interleukin-12 expression is focally enhanced in the gastric mucosa of pediatric patients with Crohn’s disease. Am J Pathol 1998;152:667–72. [PMC free article] [PubMed] [Google Scholar]
- 11.Neurath MF, Fuss I, Kelsall BL, et al. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med 1995;182:1281–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ehrhardt RO, Ludviksson BR, Gray B, et al. Induction and prevention of colonic inflammation in IL-2-deficient mice. J Immunol 1997;158:566–73. [PubMed] [Google Scholar]
- 13.Mattner F, Fischer S, Guckes S, et al. The interleukin-12 subunit p40 specifically inhibits effects of the interleukin-12 heterodimer. Eur J Immunol 1993;23:2202–8. [DOI] [PubMed] [Google Scholar]
- 14.Ling PR, Gollaher C, Colon E, et al. Human IL-12 p40 homodimer binds to the IL-12 receptor but does not mediate biologic activity. J Immunol 1995;154:116–27. [PubMed] [Google Scholar]
- 15.Gillessen S, Carvajalm D, Ling P, et al. Mouse interleukin-12 (IL-12) p40 homodimer: a potent IL-12 antagonist. Eur J Immunol 1995;25:200–6. [DOI] [PubMed] [Google Scholar]
- 16.Gately MK, Carvajal DM, Connaughton SE, et al. Interleukin-12 antagonist activity of mouse interleukin-12 p40 homodimer in vitro and in vivo. Ann N Y Acad Sci 1996;795:1–12. [DOI] [PubMed] [Google Scholar]
- 17.Camoglio L, Juffermans NP, Peppelenbosch M, et al. Contrasting roles of IL-12p40 and IL-12p35 in the development of hapten-induced colitis. Eur J Immunol 2002;32:261–9. [DOI] [PubMed] [Google Scholar]
- 18.Piccotti JR, Chan SY, Li K, et al. Differential effects of IL-12 receptor blockade with IL-12 p40 homodimer on the induction of CD4+ and CD8+ IFN-γ-producing cells. J Immunol 1997;158:643–8. [PubMed] [Google Scholar]
- 19.Magram J, Sfarra J, Connaughton S, et al. IL-12 deficient mice are defective but not devoid of type 1 cytokine responses. Ann N Y Acad Sci 1996;795:60–70. [DOI] [PubMed] [Google Scholar]
- 20.Magram J, Connaughton S, Warrier R, et al. IL-12 deficient mice are defective in IFN gamma production and type 1 cytokine responses. Immunit 1996;4:471–81. [DOI] [PubMed] [Google Scholar]
- 21.Stallmach A, Pfister K, Künne S, et al. Activation of β1 integrins mediates proliferation and inhibits apoptosis of intestinal CD4-positive lymphocytes. Eur J Immunol 2001;31:1228–38. [DOI] [PubMed] [Google Scholar]
- 22.Lieschke GJ, Rao PK, Gately MK, et al. Bioactive murine and human interleukin-12 fusion proteins which retain antitumor activity in vivo. Nat Biotechnol 1997;15:35–40. [DOI] [PubMed] [Google Scholar]
- 23.Seed B, Aruffo A. Molecular cloning of the CD2 antigen, the T-cell erythrocyte receptor, by a rapid immunoselection procedure. Proc Natl Acad Sci U S A 1987;84:3365–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boirivant M, Marini M, Di Felice G, et al. Lamina propria T cells in Crohn’s disease and other gastrointestinal inflammation show defective CD2 pathway-induced apoptosis. Gastroenterology 1999;116:557–65. [DOI] [PubMed] [Google Scholar]
- 25.Wittig B, Schwarzler C, Fohr N, et al. Curative treatment of an experimentally induced colitis by a CD44 variant V7-specific antibody. J Immunol 1998;161:1069–73. [PubMed] [Google Scholar]
- 26.Stallmach A, Wittig BM, Giese T, et al. Protection of trinitrobenzene sulfonic acid-induced colitis by an interleukin-2-IgG2b fusion protein in mice. Gastroenterology 1999;117:866–76. [DOI] [PubMed] [Google Scholar]
- 27.Barbulescu K, Becker C, Schlaak JF, et al. IL-12 and IL-18 differentially regulate the transcriptional activity of the human IFN-gamma promoter in primary CD4+ T lymphocytes. J Immunol 1998;160:3642–7. [PubMed] [Google Scholar]
- 28.Kühn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell 1993;75:263–74. [DOI] [PubMed] [Google Scholar]
- 29.Powrie F, Leach MW, Mauze S, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity 1994;1:553–62. [DOI] [PubMed] [Google Scholar]
- 30.Groux H, O’Gara A, Bigler M. A CD4+ T cell subset inhibits antigen-specific T-cell response and prevents colitis. Nature 1997;389:737–41. [DOI] [PubMed] [Google Scholar]
- 31.van Deventer SJH, Elson CO, Fedorak RN. Crohn’s Disease Study Group. 1997. Multiple doses of intravenous interleukin 10 in steroid-refractory Crohn’s disease. Gastroenterology 1997;113:383–9. [DOI] [PubMed] [Google Scholar]
- 32.Fedorak RN, Gangl A, Elson CO, et al. Recombinant human interleukin 10 in the treatment of patients with mild to moderately active Crohn’s disease. The Interleukin 10 Inflammatory Bowel Disease Cooperative Study Group. Gastroenterology 2000;119:1473–82. [DOI] [PubMed] [Google Scholar]
- 33.Schreiber S, Fedorak RN, Nielsen OH, et al. Safety and efficacy of recombinant human interleukin 10 in chronic active Crohn’s disease. Crohn’s Disease. IL-10 Cooperative Study Group. Gastroenterology 2000;119:1461–72. [DOI] [PubMed] [Google Scholar]
- 34.Colombel JF, Rutgeerts P, Malchow H, et al. 2001. Interleukin 10 (Tenovil) in the prevention of postoperative recurrence of Crohn’s disease. Gut 2001;49:42–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ina K, Itoh JK, Fukushima K, et al. Resistance of Crohn’s disease T cells to multiple apoptotic signals is associated with a Bcl-2/Bax mucosal imbalance. J Immunol 1999;163:1081. [PubMed] [Google Scholar]
- 36.Itoh J, De la Motte C, Strong SA, et al. 2001. Decreased Bax expression by mucosal T cells favours resistance to apoptosis in Crohn’s disease. Gut 2001;49:35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Targan SR, Hanauer SB, van Deventer SJ, et al. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med 1997;337:1029–35. [DOI] [PubMed] [Google Scholar]
- 38.Sandborn WJ, Feagan BG, Hanauer SB, et al. An engineered human antibody to TNF (CDP571) for active Crohn’s disease: a randomized double-blind placebo-controlled trial. Gastroenterology 2001;120:1330–8. [DOI] [PubMed] [Google Scholar]
- 39.Rutgeerts P, D’Haens G, Targan S, et al. 1999. Efficacy and safety of retreatment with anti-tumor necrosis factor antibody (infliximab) to maintain remission in Crohn’s disease. Gastroenterology 1999;117:761–9. [DOI] [PubMed] [Google Scholar]
- 40.Hanauer SB, Feagan BG, Lichtenstein GR, et al. Maintenance infliximab for Crohn’s disease: the ACCENT I randomised trial. Lancet 2002;359:1541–9. [DOI] [PubMed] [Google Scholar]
- 41.Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med 2001;345:1098–104. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Z, Correa H, Begue RE. Tuberculosis and treatment with infliximab. N Engl J Med 2002;346:623–6. [PubMed] [Google Scholar]
- 43.Holscher CAAR, Arendse B, Brown N, et al. A protective and agonistic function of IL-12p40 in mycobacterial infection. J Immunol 2001;167:6957–66. [DOI] [PubMed] [Google Scholar]
- 44.Cooper AM, Kipnis A, Turner J, et al. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular response to mycobacterial infection only if the IL-12 p40 subunit is present. J Immunol 2002;168:1322–7. [DOI] [PubMed] [Google Scholar]
- 45.Elkins KL, Cooper A, Colombini SM, et al. 2002. In vivo clearance of an intracellular bacterium, Francisella tularensis LVS, is dependent on the p40 subunit of IL-12 but not only on IL12 p70. Infect Immun 2002;70:1936–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ha SJ, Lee CH, Lee SB, et al. A novel function of IL-12p40 as a chemotactic molecule for macrophages. J Immunol 1999;163:2902–8. [PubMed] [Google Scholar]
- 47.Mullin GE, Galinkin D. Anti-IL12 imposes the death sentence on Th1 cells in TNBS colitis—is there a light at the end of the tunnel for Crohn’s disease? Inflamm Bowel Dis 2000;6:261–2. [DOI] [PubMed] [Google Scholar]
- 48.Strober W, Fuss I, Kitani A. Regulation of experimental mucosal inflammation. Acta Odontol Scand 2001;59:244–7. [DOI] [PubMed] [Google Scholar]
- 49.Zheng XX, Steele AW, Nickerson PW, et al. Administration of noncytolytic IL-10/Fc in murine models of lipopolysaccaride-induced septic shock and allogenic islet transplantation. J Immunol 1995;154:5590–600. [PubMed] [Google Scholar]