

Abstract
Background and aims: Hepatic stellate cells (HSC) are activated by liver injury to become proliferative fibrogenic myofibroblasts. This process may be regulated by the sympathetic nervous system (SNS) but the mechanisms involved are unclear.
Methods: We studied cultured HSC and intact mice with liver injury to test the hypothesis that HSC respond to and produce SNS neurotransmitters to promote fibrogenesis.
Results: HSC expressed adrenoceptors, catecholamine biosynthetic enzymes, released norepinephrine (NE), and were growth inhibited by α- and β-adrenoceptor antagonists. HSC from dopamine β-hydroxylase deficient (Dbh−/−) mice, which cannot make NE, grew poorly in culture and were rescued by NE. Inhibitor studies demonstrated that this effect was mediated via G protein coupled adrenoceptors, mitogen activated kinases, and phosphatidylinositol 3-kinase. Injury related fibrogenic responses were inhibited in Dbh−/− mice, as evidenced by reduced hepatic accumulation of α-smooth muscle actin+ve HSC and decreased induction of transforming growth factor β1 (TGF-β1) and collagen. Treatment with isoprenaline rescued HSC activation. HSC were also reduced in leptin deficient ob/ob mice which have reduced NE levels and are resistant to hepatic fibrosis. Treating ob/ob mice with NE induced HSC proliferation, upregulated hepatic TGF-β1 and collagen, and increased liver fibrosis.
Conclusions: HSC are hepatic neuroglia that produce and respond to SNS neurotransmitters to promote hepatic fibrosis.
Keywords: sympathetic nervous system, neurotransmitters, liver, fibrogenesis, hepatic stellate cells
Liver injury activates hepatic stellate cells (HSC) to move from a quiescent phenotype to a proliferative, fibrogenic, myofibroblastic phenotype.1 These activated myofibroblastic cells are responsible for the progressive accumulation of collagen that occurs as injured livers become cirrhotic. HSC are also contractile and may therefore contribute to portal hypertension.2 The mechanisms that initiate and perpetuate the fibrogenic response in injured livers are not understood.
Sympathetic nervous system (SNS) inhibitors markedly reduce experimentally induced liver fibrosis,3 and the spontaneously hypertensive rat, which has an overactive SNS, develops unusually severe liver fibrosis when given hepatotoxins.4,5 Although these observations suggest that SNS activity promotes hepatic fibrosis, the cellular target of the SNS in mediating these effects, and the mechanisms involved, are not known.
HSC may be targets for SNS regulation because we showed that cultured HSC proliferate and express collagen mRNA in response to SNS neurotransmitters.6,7 Given that HSC also express stereotypical neuroglial proteins,2,8–11 possess synaptic vesicles,10 and are innervated by autonomic fibres,12,13 we hypothesise that HSC produce SNS neurotransmitters and function as an effector arm of the SNS, automodulating their own activation via SNS neurotransmitters.
METHODS
Drugs
All drugs were from Sigma (St Louis, Missouri, USA), except pertussis toxin, wortmannin, SB202190, PD98059, and RO-32-0432 (all purchased from Calbiochem, San Diego, California, USA).
Animal experiments
Adult (10–18 weeks), male, lean, and ob/ob C57BL/6 mice were from Jackson Laboratory (Bar Harbor, Maine, USA). Similarly aged male Sprague-Dawley rats were from Charles River (Wilmington, Massachusetts, USA). Male dopamine β-hydroxylase deficient (Dbh−/−) and Dbh+/− mice (30–40 weeks) were from our colony.14 Most animals were used to provide HSC for in vitro experiments (see below). However, other experiments were done in intact mice. In those studies, Dbh−/− mice (n = 12) and Dbh+/− littermates (n = 6) were fed methionine restricted choline deficient (MCD) diets (ICN, Aurora, Ohio, USA) for four weeks to induce steatohepatitis and liver fibrosis.15,16 Half of these Dbh−/− mice were given subcutaneous osmotic minipumps (Alzet, Cupertino, California, USA) that infused isoprenaline at 20 mg/kg/day to induce SNS activation.17 Some ob/ob mice (n = 15) were similarly infused with vehicle or norepinephrine (NE 2.5 mg/kg/day). Such NE treatment induces SNS activation in normal mice.18 During in vivo experiments, mice were weighed weekly until sacrifice when livers were harvested, fixed (in buffered formalin or optimal cutting temperature fixative (Sakura, Torrance, California, USA)), or snap frozen and stored at −80°C. All experiments satisfied the guidelines of our Institutional Animal Care Committee and the National Institutes of Health.
Stellate cell isolation and culture
Pronase-collagenase liver digestion was used to isolate HSC from wild type, Dbh+/−, and Dbh−/− mice, or healthy rats. For each experiment, HSC were pooled from six mice of each genotype. A single rat provided sufficient HSC for an experiment. All experiments were replicated at least twice. In total, HSC were obtained from about 72 mice and two rats. Cell identity was confirmed by autofluorescence and expression of alpha smooth muscle actin (ASMA) and glial fibrillary acidic protein (GFAP).7 HSC preparation purity was always >95%.7 HSC were routinely cultured in 10% serum supplemented RPMI 1640 medium. To assess catecholamine production, HSC and conditioned media were harvested after four days in culture; cells were washed twice with ice cold phosphate buffered saline. Cells or conditioned medium were then rapidly analysed for catecholamine content by high pressure liquid chromatography (HPLC).
Apoptosis assays
Equal numbers of HSC were plated on 6 mm petri dishes with or without 10 μM prazosin. Fresh prazosin was added every two days when the medium was changed. At harvest, apoptotic activity was assessed with the Vybrant (annexin V) apoptosis assay kit (Molecular Probes, Eugene, Oregon, USA).
Cell proliferation assay
Freshly isolated HSC were seeded into culture flasks, grown to subconfluence (~day 7), and then harvested, resuspended in serum free medium, and re-plated on 96 well plates at 5000 cells/well. Twenty four hours later, when the cells had become quiescent, NE (10 μM) with or without various inhibitors (for example, prazosin (10 μM), pertussis toxin (100 ng/ml), wortmannin (100 nM), SB202190 (10 μm), PD98059 (20 μm), or RO-32-0432 (1 μm)) were added to some wells.6,19–26 After 44 hours, cell numbers were assessed by a further four hour incubation with WST-8 tetrazolium reagent (Dojindo Molecular Technologies, Gaithersburg, Maryland, USA).7,19,27 In viable cells, the tetrazolium salt is metabolised to a colorimetric dye and cell number is proportional to the signal intensity at 450 nm.7 As detailed in information that the manufacturer provides with the reagents, the use of tetrazolium dyes has been validated against thymidine incorporation for a variety of cell types, including stellate cells.19,27–30 Therefore, we7 and others19 have used this assay to evaluate HSC proliferation.
RT-PCR
RNA was extracted from HSC using RNeasy kits (Qiagen, Valencia, California, USA). One step reverse transcription-polymerase chain reaction (RT-PCR) was performed with Superscript one step RT-PCR with platinum Taq kits (Invitrogen, Carlsbad, California, USA) and Ambion’s QuantumRNA Classic II 18S internal standard (Ambion, Austin, Texas, USA). Products were separated by electrophoresis on a 1.5% agarose gel using published primer sequences and conditions.20,31
HPLC analysis
Catecholamines were extracted from HSC pellets and HSC conditioned medium with perchloric acid. NE, dopamine, 3,4-dihydroxyphenylacetic acid (DOPAC), serotonin (5-hydroxytryptamine, 5-HT), 5-hydroxyindoleacetic acid (5-HIAA), and homovallinic acid (HVA) were analysed by HPLC,32 using dihydroxybenzilamine as the internal standard. Catecholamine concentrations were calculated with a Hewlett Packard integrator.
Immunoblot
Net protein content of HSC homogenates was quantified by BSA assay (Pierce, Rockford, Illinois, USA) using bovine serum albumin standards. Proteins (10 μg/lane) were then resolved by polyacrylamide gel electrophoresis and transferred to nylon membranes. After membranes were incubated with primary antibodies to ASMA (1:3000; Sigma), tyrosine hydroxylase (TH 1:100; Novus Biologics, Littleton, Colorado, USA), dopamine beta-hydroxylase (1:250; Research Diagnostics, Flanders, New Jersey, USA), α-1A, α-1B, and α-1D adrenoceptors (1:100; Santa Cruz Biotech, Santa Cruz, California, USA), and β1, β2, and β3 adrenoceptors (1:200; Santa Cruz Biotech), peroxidase conjugated secondary antibodies were added. Antigens were demonstrated by enhanced chemiluminescence (Amersham Biosciences, Piscataway, New Jersey, USA).33
Immunohistochemistry
After being deparaffinised, rehydrated, and heated in a microwave oven for 10 minutes at 750 Watt in citrate buffer, pH 6.0, liver sections were incubated at room temperature for 30 minutes with primary antibodies. Mouse monoclonal anti-ASMA antibody (1/40 dilution; Dako, Denmark) and rabbit polyclonal anti-GFAP (1/300 dilution; Dako) were used to identify either activated HSC1 or both activated and quiescent HSC,11 respectively. Primary antibody binding was revealed with the peroxidase animal research kit (Dako).
Histology
Formalin fixed paraffin embedded liver sections were stained Masson trichrome. Coded samples were examined. The degree of fibrosis was scored as 0 = no fibrosis; 1 = mild fibrosis; 2 = moderate fibrosis; and 3 = severe fibrosis.
RNA isolation and ribonuclease protection assay
Total RNA was isolated from liver samples.34,35 Collagen-1-α1, transforming growth factor β1 (TGF-β1), matrix metalloproteinase (MMP)-2 and -9, and tissue inhibitor of metalloproteinase (TIMP)-1 and -2 expression were evaluated by ribonuclease protection assay kits (PharMingen, San Diego, California, USA).35
Statistics
Statistical analysis by the unpaired t test or the Mann-Whitney test was performed with Graphpad Prism software (San Diego, California, USA). Significance was accepted as p<0.05.
RESULTS
HSC express catecholamine biosynthetic enzymes
Expression of key enzymes in the catecholamine biosynthetic pathway was evaluated in primary HSC cultured from normal mice. Both TH and dopamine β-hydroxylase (Dbh) were demonstrated (fig 1 ▶).
Figure 1.

Expression of tyrosine hydroxylase and dopamine β-hydroxylase by cultured hepatic stellate cells (HSC). HSC from six healthy adult mice were pooled and cultured for four days. Lysates were evaluated by immunoblot (10 μg protein/lane).
Primary HSC synthesise and release NE in culture
HPLC analysis of HSC conditioned medium from normal mice HSC showed that these cells also released NE (69 (6) ng/ml). In contrast, NE was not detected in conditioned medium from Dbh−/− HSC or in unconditioned medium that had not been exposed to HSC. In addition to NE, normal murine HSC lysates contained dopamine, 5-HT, catecholamine metabolites (DOPAC and HVA), and the 5-HT metabolite 5-HIAA (fig 2 ▶).
Figure 2.

Primary hepatic stellate cell (HSC) synthesise and release of norepinephrine (NE) in culture. HSC pooled from six normal mice were cultured for four days. Cell lysates and conditioned media were analysed by high pressure liquid chromatography. DOPAC, 3,4-dihydroxyphenylacetic acid; DBA, dihydroxybenzilamine; DA, dopamine; 5-HIAA, 5-hydroxyindoleacetic acid; HVA, homovallinic acid; 5-HT, 5-hydroxytryptamine.
HSC express multiple adrenoceptor subtypes
Although HSC are known to express α1-adrenoceptors,36 it is not known which α-adrenoceptors subtypes are expressed, or if HSC express β-adrenoceptors. RT-PCR analysis demonstrated that primary HSC expressed α-1B, α-1D, β1, and β2 adrenoceptors (fig 3 ▶). A similar expression profile was noted with western blot analysis (not shown).
Figure 3.

Hepatic stellate cells (HSC) express multiple adrenoceptor subtypes. RNA obtained by pooling HSC from six normal mice was analysed by reverse transcription-polymerase chain reaction. First lane, DNA ladder (500–200 bp, arrowed). Each subsequent pair of lanes is a replicate analysis of adrenoceptor genes. The 18S band (324 bp) serves as a control.
Adrenoceptor antagonists inhibit the growth of primary HSC cultures
To determine if endogenous NE is important for HSC growth, we plated wild-type HSC in the presence and absence of the α1-adrenoceptor antagonist prazosin (10 μM) and the β-adrenoceptor antagonist propranolol (10 μM). Prazosin and propranolol each reduced HSC numbers by ~20%. Moreover, the combination of prazosin and propranolol decreased HSC growth by ~50%. Thus the growth inhibitory actions of the α- and β-adrenoceptor antagonists were additive (fig 4 ▶).
Figure 4.

Adrenoceptor antagonists inhibited the growth of primary hepatic stellate cell (HSC) cultures. Freshly isolated HSC pooled from six normal mice were cultured with prazosin (PRZ 10 μM) and propranolol (PRL 10 μM) alone and PRZ+PRL (PP). After 48 hours, cell numbers were evaluated. Data (mean (SD)) are from two separate experiments. *p<0.05 for PRZ or PRL treated HSC versus control, ††p<0.01 for PP treated HSC versus control.
HSC that are genetically incapable of producing NE grow poorly in culture and exogenous NE rescues proliferative activity
To confirm that NE is an autocrine growth factor for HSC, we studied HSC cultured from Dbh−/− (NE deficient) and Dbh+/− (control) mice. The same numbers of cells were plated from each group. To assure that the culture conditions permitted NE activity, some HSC from Dbh+/− mice were also incubated with prazosin (10 μM). During four days in culture, HSC from Dbh+/− mice proliferated to become nearly confluent (fig 5A ▶). This proliferative activity was inhibited significantly by prazosin (fig 5B ▶). Proliferative activity was also significantly reduced in HSC from Dbh−/− mice (fig 5C ▶). Exogenous NE rescued the growth of Dbh−/− HSC (fig 5D ▶), confirming the importance of endogenous NE for HSC growth.
Figure 5.

Hepatic stellate cells (HSC) that are genetically incapable of producing norepinephrine (NE) grow poorly in culture and exogenous NE rescues proliferative activity. Representative photomicrographs of pooled HSC isolated from six Dbh+/− mice cultured without (A) or with (B) prazosin (PRZ 10 μM) for four days, and four day old cultures of HSC pooled from six control Dbh−/− mice (C). HSC from six additional Dbh−/− mice were cultured in control medium or medium+NE (10 μM) for four days and HSC numbers were quantified (D). *p<0.05 versus control.
NE is an autocrine growth factor for HSC
Normal HSC become spontaneously activated during culture and proliferate at a greater rate than they die. Therefore, increases in proliferative activity normally drive HSC growth in culture. To determine to what extent, if any, NE related differences in cell number might also reflect differences in apoptotic activity, HSC were harvested, incubated with annexin V, and analysed by flow cytometry. After one day in culture, a slightly greater proportion of apoptotic HSC were detected in cultures from Dbh−/− mice (12.4 (0.26)%) compared with Dbh+/− mice (9.96 (0.15)%) (p = 0.007). Therefore, endogenous NE promotes the viability of HSC. This antiapoptotic effect may help to explain why endogenous NE is required for optimal HSC growth.
NE activates adrenoceptor G protein coupled mechanisms that induce mitogenic and survival pathways
Previously, we showed that adding NE to cultures of murine HSC caused a dose related increase in proliferation that was blocked by coadministration of prazosin.6 To be certain that this response was not an unusual property of murine HSC, we cultured rat HSC with NE, with and without prazosin. Incubation with NE for 48 hours significantly (p<0.05) increased rat HSC numbers, and prazosin significantly (p<0.05) blocked these trophic effects, demonstrating that NE generally promotes HSC proliferation.
We next addressed potential mechanisms for this effect. Adrenoceptors are known to be coupled to G proteins in other mesenchymal cells in which they induce proliferation by activating phosphoinositol triphosphate and mitogen activated protein kinases.20,21,24 Therefore, primary HSC from normal mice were cultured with NE in the presence and absence of pertussis toxin, a G protein inhibitor; wortmannin, a PI3-kinase inhibitor; SB202190, an inhibitor of p38 MAP kinase; PD98059, a MEK inhibitor; and Ro-32-0432, a protein kinase C inhibitor. Pertussis toxin, wortmannin, and PD98059 significantly reduced NE induced proliferation (fig 6 ▶). Thus the proliferative effects of NE on HSC are mediated by G protein coupled receptors and the downstream effects involve PI-3 kinase and the Erk family of mitogen activated protein kinases.
Figure 6.

Norepinephrine (NE) activates adrenoceptor G protein coupled mechanisms that induce mitogenic and survival pathways in hepatic stellate cells (HSC). HSC pooled from six normal mice were cultured with NE (10 μM) or NE with pertussis toxin (NE+PT), wortmannin (NE+WT), PD98059 (NE+PD), SB202190 (NE+SB), or Ro-32-0432 (NE+RO). After two days, HSC numbers were evaluated in triplicate wells. Mean (SEM) results of duplicate experiments are shown. *p<0.05 for inhibitor treated versus NE alone.
Adrenergic agonists regulate HSC activation in intact mice
To determine if NE had any impact on HSC function in intact animals, NE was infused chronically into ob/ob mice, which have low levels of NE37,38 and are resistant to fibrosis despite having chronic steatohepatitis.16,39,40 Immunohistochemistry was used to demonstrate GFAP(+) cells in livers because this marker provides a reliable estimate of both quiescent and activated HSC.41 Control ob/ob mice have significantly fewer HSC than their lean littermates. Continuous subcutaneous infusion of NE for four weeks markedly stimulated HSC proliferation in ob/ob mice such that the numbers of GFAP(+) HSC in NE treated ob/ob mice doubled, leading to HSC numbers that approached those of their control littermates (fig 7 ▶).
Figure 7.

Norepinephrine (NE) increases hepatic stellate cell (HSC) activation in NE deficient ob/ob mice. Ob/ob mice were treated with NE (n = 5) or vehicle (n = 10). After four weeks, glial acidic fibrillary protein positive cells were counted in five randomly selected high power fields/liver section from each animal. Data are mean (SD). *p<0.05 for ob/ob control versus lean control; †p<0.05 for ob/ob+NE versus ob/ob control.
Ob/ob mice are genetically incapable of producing leptin, a factor that is known to activate HSC proliferation by direct interaction with HSC receptors.19,42 Therefore, it is conceivable that the effects of NE in this strain might have been confounded by leptin deficiency. To exclude this possibility, we studied Dbh−/− mice, which also have reduced NE but are not leptin deficient. We fed Dbh−/− mice and Dbh+/− mice antioxidant depleted diets. Normal mice develop steatohepatitis and fibrosis after eating these diets for 4–8 weeks.16 Consistent with this literature, after four weeks of treatment, control Dbh+/− mice exhibit striking accumulation of HSC that express ASMA, an accepted marker of HSC activation (fig 8A ▶, B). In contrast, ASMA+ve HSC cannot be demonstrated in Dbh−/− mice (fig 8A ▶, C). This finding is unlikely to be a staining artefact because ASMA+ve vessel walls are easily identified (fig 8C ▶). Moreover, compared with Dbh+/− controls, Dbh−/− mice exhibit significantly less hepatic expression of TGF-β1 and collagen, two other indicators of HSC activation (fig 9A ▶, B). Ribonuclease protection analysis also demonstrated a small (~30–40%) but statistically significant (p<0.05) reduction in induction of TIMP-2 transcripts in the Dbh−/− group. The diet barely increased expression of other genes (for example, MMP-2, MMP-9, and TIMP-1) that regulate matrix degradation, and therefore no group related differences in these transcripts were observed.
Figure 8.

Figure 9.
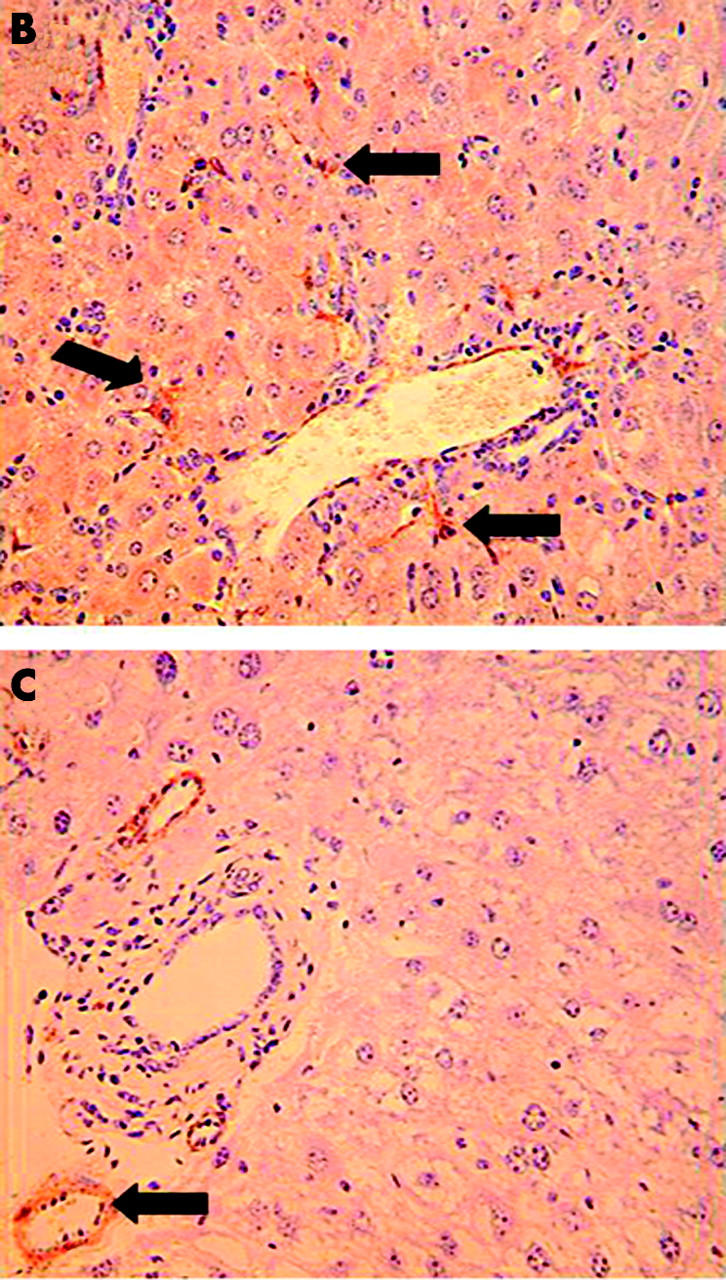
Reduced activation of hepatic stellate cells (HSC) in norepinephrine (NE) deficient Dbh−/− mice. Twelve Dbh−/− and six of their control Dbh+/− littermates were fed methionine choline deficient (MCD) diets. Half of the Dbh−/− mice were also infused with isoprenaline (ISO) for four weeks. (A) Alpha smooth muscle actin (ASMA)+ve sinusoidal cells were counted in five randomly selected fields/liver section from each mouse. Mean (SD) results of one experiment are shown. *p<0.05 for Dbh+/− versus Dbh−/− control; †p<0.05 for Dbh−/− control versus Dbh−/− +ISO. Identical results have since been obtained in a second experiment that studied an additional 12 mice (four mice/group). (B) Photomicrograph from representative Dbh+/− mice. (C) Photomicrograph from typical Dbh−/− mice. Arrows indicate typical ASMA+ve HSC (B) or vessel wall (C).
To verify that it was reduced adrenoceptor activity that prevented HSC activation, we implanted osmotic minipumps that contained vehicle or isoprenaline, a β-adrenoceptor agonist, into Dbh−/− mice and repeated the feeding experiment. Infusion of isoprenaline rescued HSC activation in Dbh−/− mice and returned numbers of ASMA (+) HSC to levels exhibited by Dbh+/− mice that were also fed the hepatotoxic diet (fig 8A ▶). Isoprenaline infusion similarly normalised induction of TGF-β in Dbh−/− mice (fig 9B ▶). In parallel experiments, we examined the effect of NE, an α-adrenoceptor agonist, on HSC activation in ob/ob mice. Compared with control ob/ob mice, ob/ob mice that were treated with NE minipumps for four weeks had significantly increased liver expression of TGF-β1 and collagen mRNA (fig 10A ▶, B). Indeed, in two of five ob/ob mice, NE treatment increased hepatic fibrosis sufficiently for scarring to be detected simply by staining liver sections with Mason trichrome reagent (fig 10C ▶). No such fibrosis was detected in twice as many ob/ob controls. Therefore, both α-predominant (NE, fig 7 ▶) and β-predominant (isoprenaline, figs 8A ▶, 9B ▶) adrenoceptor agonists affect HSC activation in vivo. Despite this increase in HSC activation, alanine aminotransferase values in NE treated ob/ob mice were lower than their littermate controls (fig 10D ▶). Therefore, NE related increases in fibrogenesis are not easily attributed to NE exacerbation of liver injury in leptin deficient mice.
Figure 10.
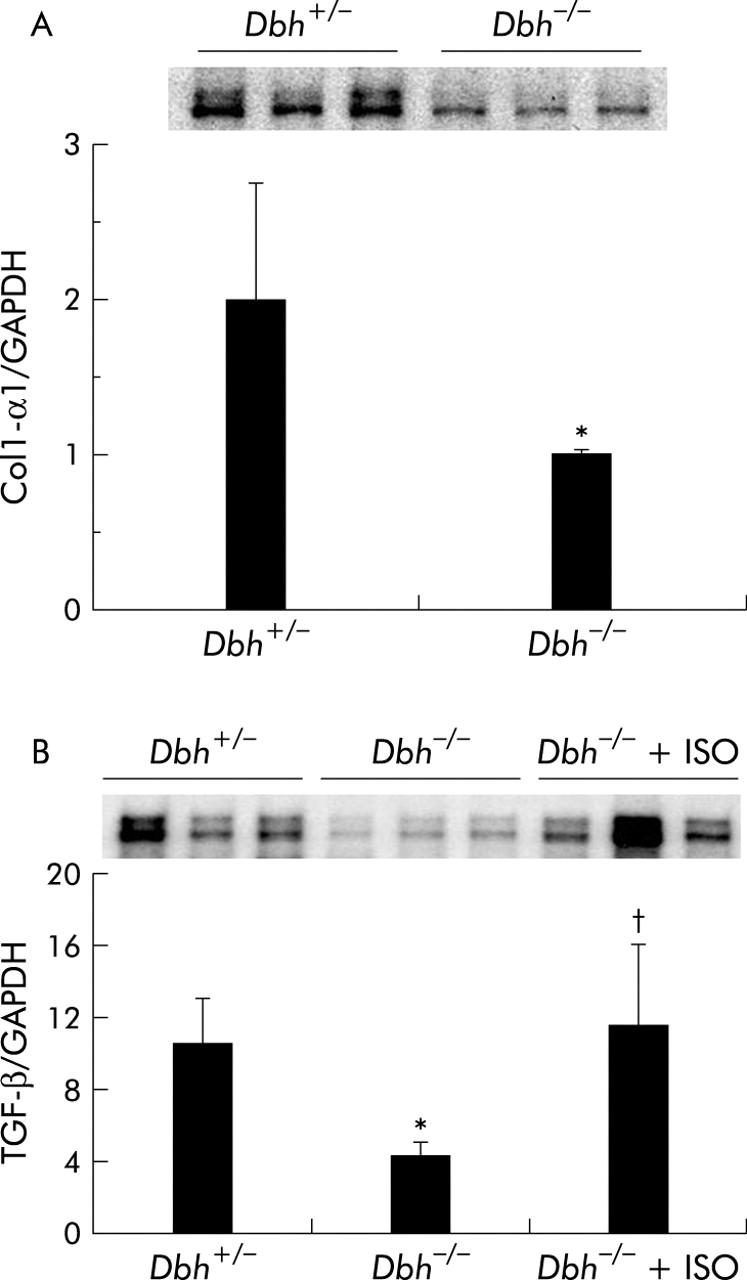
Norepinephrine (NE) regulates hepatic expression of collagen and transforming growth factor β1 (TGF-β). Liver RNA was isolated from six Dbh+/− mice, six Dbh−/− mice, and six Dbh−/− mice that were infused with isoprenaline (ISO). All mice had been fed methionine choline deficient (MCD) diets for four weeks. Hepatic expression of collagen-1-α1 (Col1-α1) (A) and TGF-β (B) was evaluated by ribonuclease protection assay (20 μg RNA/assay). A representative phosphoimage displays individual data from three mice/group from the first ribonuclease protection assay. Normalised mean (SD) collagen (n = 12; six mice/group) and TGF-β1 gene expression from all 18 mice (six mice/group) are shown. *p<0.05 for Dbh+/− versus Dbh−/− mice; †p<0.05 for Dbh−/− versus Dbh−/−+ISO.
DISCUSSION
We have shown that HSC express key enzymes for catecholamine biosynthesis, and produce NE and other catecholamines. Moreover, HSC use catecholamines to autoregulate their growth because increases in HSC number are significantly attenuated by culturing normal HSC with α- or β-adrenoceptor antagonists. Inhibitor studies suggest that the trophic actions of NE require activation of kinases that promote cell viability and proliferation. This finding is intriguing because leptin, another factor that promotes HSC growth, was recently shown to activate similar signalling mechanisms.43,44 Pertinent to the latter report, our results demonstrate that HSC numbers and activation are reduced in leptin deficient mice which are known to have reduced levels of NE38 and a severely impaired fibrogenic response to liver injury.16,40 In addition, we found that NE treatment normalised HSC numbers and activation parameters despite persistent leptin deficiency. Together, these findings suggest that NE lies downstream of leptin in the process of HSC activation. We did not evaluate whether or not leptin induces production of NE by HSC, as it is known to do in adipocytes.45 It is also conceivable that leptin promotes HSC activation by inducing expression or function of adrenoreceptors or components of the post-receptor signalling pathways that mediate NE effects. More work is required to delineate the exact nature of leptin-NE interactions. Nevertheless, our findings in Dbh−/− mice, which produce leptin and have normal leptin receptors but are deficient in NE,46 support the general importance of adrenoceptor agonists in HSC activation. In these mice that are genetically incapable of converting dopamine into NE, hepatotoxic diets only activate HSC when adrenoceptor activity is increased by treatment with adrenergic agents. Thus when NE is deficient, leptin cannot fully activate HSC, and TGF-β1 and collagen are not induced normally in response to liver injury.
That NE may subserve functions other than its classically assigned role of neurotransmission is becoming well established in other organs. For example, cardiac remodelling in heart failure involves mitogenic and fibrogenic actions of NE that are mediated via adrenoceptors.24,47,48 The present studies provide novel evidence that HSC are both a source and a direct cellular target of NE. This extends our understanding of the mechanisms that mediate the profibrogenic actions of catecholamines in the liver and suggests that targeted interruption of catecholamine signalling in hepatic stellate cells may be a useful therapeutic approach to constrain the fibrogenic response to liver injury.
Figure 11.

Figure 12.

Figure 13.

Norepinephrine (NE) increases hepatic expression of collagen and transforming growth factor β1 (TGF-β) without increasing liver injury in NE deficient ob/ob mice. Liver RNA was isolated from five control ob/ob mice and five NE treated ob/ob mice. In each mouse, hepatic expression of collagen-1-α1 (Col1-α1) (A) and TGF-β (B) was evaluated by ribonuclease protection assay (20 μgRNA/assay). A representative phosphoimage demonstrating individual data from two animals per group is shown. Normalised mean (SD) collagen and TGF-β1 gene expression in all mice are shown. *p<0.05 control ob/ob versus ob/ob+NE. (C) Mason-trichrome stained sections from a representative control ob/ob mouse and from an NE treated ob/ob mouse with pericellular and sinusoidal fibrosis. (D) Mean (SEM) alanine aminotransferase (ALT) values in the control and NE treated groups of ob/ob mice. *p<0.05 for NE treated ob/ob versus ob/ob controls.
Acknowledgments
This work was supported by NIAAA RO1-10154 (AMD), NIAAA RO1-12059 (AMD), and FWO Vlaanderen G.0139.00N (TR).
Abbreviations
ASMA, alpha smooth muscle actin
Dbh, dopamine β-hydroxylase
DOPAC, 3,4-dihydroxyphenylacetic acid
GFAP, glial acidic fibrillary protein
HPLC, high pressure liquid chromatography
HSC, hepatic stellate cells
HVA, homovallinic acid
MCD, methionine restricted choline deficient
MMP, matrix metalloproteinase
NE, norepinephrine
5-HT, 5-hydroxytryptamine (serotonin)
RT-PCR, reverse transcription-polymerase chain reaction
SNS, sympathetic nervous system
TGF-β1, transforming growth factor β1
TH, tyrosine hydroxylase
TIMP, tissue inhibitor of metalloproteinase
5-HIAA, 5-hydroxyindoleacetic acid
REFERENCES
- 1.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000;275:2247–50. [DOI] [PubMed] [Google Scholar]
- 2.Reynaert H, Thompson MG, Thomas T, et al. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut 2002;50:571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubuisson L, Desmouliere A, Decourt B, et al. Inhibition of rat liver fibrogenesis through noradrenergic antagonism. Hepatology 2002;35:325–31. [DOI] [PubMed] [Google Scholar]
- 4.Hsu CT. The role of the sympathetic nervous system in promoting liver cirrhosis induced by carbon tetrachloride, using the essential hypertensive animal (SHR). J Auton Nerv Syst 1992;37:163–73. [DOI] [PubMed] [Google Scholar]
- 5.Hsu CT. The role of the autonomic nervous system in chemically-induced liver damage and repair-using the essential hypertensive animal model (SHR). J Auton Nerv Syst 1995;51:135–42. [DOI] [PubMed] [Google Scholar]
- 6.Oben JA, Yang S, Lin H, et al. Norepinephrine and neuropeptide Y promote proliferation and collagen gene expression of hepatic myofibroblastic stellate cells. Biochem Biophys Res Commun 2003;302:685–90. [DOI] [PubMed] [Google Scholar]
- 7.Oben JA, Yang S, Lin H, et al. Acetylcholine promotes the proliferation and collagen gene expression of myofibroblastic hepatic stellate cells. Biochem Biophys Res Commun 2003;300:172–7. [DOI] [PubMed] [Google Scholar]
- 8.Buniatian G, Hamprecht B, Gebhardt R. Glial fibrillary acidic protein as a marker of perisinusoidal stellate cells that can distinguish between the normal and myofibroblast-like phenotypes. Biol Cell 1996;87:65–73. [PubMed] [Google Scholar]
- 9.Niki T, Pekny M, Hellemans K, et al. Class VI intermediate filament protein nestin is induced during activation of rat hepatic stellate cells. Hepatology 1999;29:520–7. [DOI] [PubMed] [Google Scholar]
- 10.Cassiman D, van Pelt J, De Vos R, et al. Synaptophysin: A novel marker for human and rat hepatic stellate cells. Am J Pathol 1999;155:1831–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cassiman D, Denef C, Desmet VJ, et al. Human and rat hepatic stellate cells express neurotrophins and neurotrophin receptors. Hepatology 2001;33:148–58. [DOI] [PubMed] [Google Scholar]
- 12.Bioulac-Sage P, Lafon ME, Saric J, et al. Nerves and perisinusoidal cells in human liver. J Hepatol 1990;10:105–12. [DOI] [PubMed] [Google Scholar]
- 13.Ueno T, Sata M, Sakata R, et al. Hepatic stellate cells and intralobular innervation in human liver cirrhosis. Hum Pathol 1997;28:953–9. [DOI] [PubMed] [Google Scholar]
- 14.Thomas SA, Palmiter RD. Impaired maternal behavior in mice lacking norepinephrine and epinephrine. Cell 1997;91:583–92. [DOI] [PubMed] [Google Scholar]
- 15.Akhurst B, Croager EJ, Farley-Roche CA, et al. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology 2001;34:519–22. [DOI] [PubMed] [Google Scholar]
- 16.Leclercq IA, Farrell GC, Schriemer R, et al. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol 2002;37:206–13. [DOI] [PubMed] [Google Scholar]
- 17.Mackintosh CA, Feith DJ, Shantz LM, et al. Overexpression of antizyme in the hearts of transgenic mice prevents the isoprenaline-induced increase in cardiac ornithine decarboxylase activity and polyamines, but does not prevent cardiac hypertrophy. Biochem J 2000;350:645–53. [PMC free article] [PubMed] [Google Scholar]
- 18.Vecchione C, Fratta L, Rizzoni D, et al. Cardiovascular influences of (alpha)1b-adrenergic receptor defect in mice. Circulation 2002;105:1700–7. [DOI] [PubMed] [Google Scholar]
- 19.Saxena NK, Yang Y, Floyd J, et al. Leptin is mitogenic, anti-apoptotic and increases fibrogenic response genes in rat hepatic stellate cells. Hepatology 2002;36:316A. [Google Scholar]
- 20.Hakuno D, Fukuda K, Makino S, et al. Bone marrow-derived regenerated cardiomyocytes (CMG cells) express functional adrenergic and muscarinic receptors. Circulation 2002;105:380–6. [DOI] [PubMed] [Google Scholar]
- 21.Zou Y, Komuro I, Yamazaki T, et al. Both Gs and Gi proteins are critically involved in isoproterenol-induced cardiomyocyte hypertrophy. J Biol Chem 1999;274:9760–70. [DOI] [PubMed] [Google Scholar]
- 22.Tangkijvanich P, Santiskulvong C, Melton AC, et al. p38 MAP kinase mediates platelet-derived growth factor-stimulated migration of hepatic myofibroblasts. J Cell Physiol 2002;191:351–61. [DOI] [PubMed] [Google Scholar]
- 23.Ajizian SJ, English BK, Meals EA. Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-gamma. J Infect Dis 1999;179:939–44. [DOI] [PubMed] [Google Scholar]
- 24.Xiao L, Pimental DR, Amin JK, et al. MEK1/2-ERK1/2 mediates alpha1-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J Mol Cell Cardiol 2001;33:779–87. [DOI] [PubMed] [Google Scholar]
- 25.Hansel DE, Eipper BA, Ronnett GV. Neuropeptide Y functions as a neuroproliferative factor. Nature 2001;410:940–4. [DOI] [PubMed] [Google Scholar]
- 26.Wu H-L, Albrightson C, Nambi P. Selective inhibition of rat mesangial cell proliferation by a synthetic peptide derived from the sequence of the C2 region of PKC[beta]. Peptides 1999;20:675–8. [DOI] [PubMed] [Google Scholar]
- 27.Frank S, Stallmeyer B, Kampfer H, et al. Leptin enhances wound re-epithelialization and constitutes a direct function of leptin in skin repair. J Clin Invest 2000;106:501–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983;65:55–63. [DOI] [PubMed] [Google Scholar]
- 29.Isobe I, Michikawa M, Yanagisawa K. Enhancement of MTT, a tetrazolium salt, exocytosis by amyloid beta-protein and chloroquine in cultured rat astrocytes. Neurosci Lett 1999;266:129–32. [DOI] [PubMed] [Google Scholar]
- 30.Matsuoka M, Wispriyono B, Igisu H. Increased cytotoxicity of cadmium in fibroblasts lacking c-fos. Biochem Pharmacol 2000;59:1573–6. [DOI] [PubMed] [Google Scholar]
- 31.Hutchinson DS, Evans BA, Summers RJ. (beta)1-Adrenoceptors compensate for (beta)3-adrenoceptors in ileum from (beta)3-adrenoceptor knock-out mice. Br J Pharmacol 2001;132:433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ladenheim B, Krasnova IN, Deng X, et al. Methamphetamine-induced neurotoxicity is attenuated in transgenic mice with a null mutation for interleukin-6. Mol Pharmacol 2000;58:1247–56. [DOI] [PubMed] [Google Scholar]
- 33.Koteish A, Yang S, Lin H, et al. Chronic ethanol exposure potentiates lipopolysaccharide liver injury despite inhibiting Jun N-terminal kinase and caspase 3 activation. J Biol Chem 2002;277:13037–44. [DOI] [PubMed] [Google Scholar]
- 34.Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156–9. [DOI] [PubMed] [Google Scholar]
- 35.Elenkov IJ, Chrousos GP, Wilder RL. Neuroendocrine regulation of IL-12 and TNF-alpha/IL-10 balance. Clinical implications. Ann NY Acad Sci U S A 2000;917:94–105. [DOI] [PubMed] [Google Scholar]
- 36.Athari A, Hanecke K, Jungermann K. Prostaglandin F2 alpha and D2 release from primary Ito cell cultures after stimulation with noradrenaline and ATP but not adenosine. Hepatology 1994;20:142–8. [DOI] [PubMed] [Google Scholar]
- 37.Knehans AW, Romsos DR. Reduced norepinephrine turnover in brown adipose tissue of ob/ob mice. Am J Physiol 1982;242:E253–61. [DOI] [PubMed] [Google Scholar]
- 38.Young JB, Landsberg L. Diminished sympathetic nervous system activity in genetically obese (ob/ob) mouse. Am J Physiol 1983;245:E148–54. [DOI] [PubMed] [Google Scholar]
- 39.Ikejima K, Honda H, Yoshikawa M, et al. Leptin augments inflammatory and profibrogenic responses in the murine liver induced by hepatotoxic chemicals. Hepatology 2001;34:288–97. [DOI] [PubMed] [Google Scholar]
- 40.Honda H, Ikejima K, Hirose M, et al. Leptin is required for fibrogenic responses induced by thioacetamide in the murine liver. Hepatology 2002;36:12–21. [DOI] [PubMed] [Google Scholar]
- 41.Cassiman D, Libbrecht L, Desmet V, et al. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol 2002;36:200–9. [DOI] [PubMed] [Google Scholar]
- 42.Ikejima K, Takei Y, Honda H, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology 2002;122:1399–410. [DOI] [PubMed] [Google Scholar]
- 43.Titus MA, Saxena NK, Floyd JJ, et al. Leptin signal transduction in hepatic stellate cells involves activation of the mitogen activated protein kinase (MAPK) p44/42 or ERK, as well as the signal transducer and activator of transcription 3 (Stat 3). Gastroenterology 2003;124:A634. [Google Scholar]
- 44.Ikejima K, Lang T, Yoshikawa M, et al. Leptin enhances PDGF-dependent cell growth in hepatic stellate cells: involvement of the PI3K-AKT pathway. Gastroenterology 2003;254:A254. [Google Scholar]
- 45.Commins SP, Marsh DJ, Thomas SA, et al. Norepinephrine is required for leptin effects on gene expression in brown and white adipose tissue. Endocrinology 1999;140:4772–8. [DOI] [PubMed] [Google Scholar]
- 46.Thomas SA, Palmiter RD. Thermoregulatory and metabolic phenotypes of mice lacking noradrenaline and adrenaline. Nature 1997;387:94–7. [DOI] [PubMed] [Google Scholar]
- 47.Akiyama-Uchida Y, Ashizawa N, Ohtsuru A, et al. Norepinephrine enhances fibrosis mediated by TGF-beta in cardiac fibroblasts. Hypertension 2002;40:148–54. [DOI] [PubMed] [Google Scholar]
- 48.Fisher SA, Absher M. Norepinephrine and ANG II stimulate secretion of TGF-beta by neonatal rat cardiac fibroblasts in vitro. Am J Physiol 1995;268:C910–17. [DOI] [PubMed] [Google Scholar]