

Abstract
Background: IgA class serum autoantibodies against type 2 (tissue) transglutaminase (TG2) bind to both intestinal and extraintestinal normal tissue sections in vitro, eliciting endomysial, reticulin, and jejunal antibody reactions. It is not known whether similar binding also occurs in coeliac patients in vivo, and may thereby contribute to disease manifestations.
Aims: To investigate intestinal and extraintestinal coeliac tissues for the presence of in vivo bound TG2 specific IgA and its relation to small intestinal mucosal atrophy.
Patients: We investigated jejunal samples with normal villous morphology from 10 patients with developing coeliac disease who subsequently progressed to a flat lesion, from 11 patients with dermatitis herpetiformis, and from 12 non-coeliac controls. Six extrajejunal biopsy samples (liver, lymph node, muscle, appendix), obtained based on independent clinical indications from patients with active coeliac disease, were also studied.
Methods: Double colour immunofluorescent studies for in situ IgA, TG2, and laminin were performed. IgA was eluted from tissue sections and tested for TG2 specificity by enzyme linked immunosorbent assay and indirect immunofluorescence.
Results: IgA (in one IgA deficient case IgG) deposition on extracellularly located TG2 was detected in jejunal and extrajejunal specimens of all coeliac patients, and also in seven of 11 dermatitis herpetiformis patients, of whom two had no circulating endomysial antibodies. IgA eluted from extraintestinal coeliac tissues was targeted against TG2.
Conclusions: Coeliac IgA targets jejunal TG2 early in disease development even when endomysial antibodies are not present in the circulation. Extraintestinal target sites of coeliac IgA further indicate that humoral immunity may have a pathogenetic role.
Keywords: autoantibodies, coeliac disease, dermatitis herpetiformis, immunoglobulin deposition, transglutaminase type 2, tissue transglutaminase
In a certain proportion of individuals carrying the extended autoimmune HLA DR3-DQ2 or DR4-DQ8 haplotype, daily ingested wheat, rye, and barley proteins induce small intestinal villous atrophy and crypt hyperplasia, resulting in a symptomatic gastrointestinal disease.1,2 Clinical research has also focused on gluten induced entities with no or mild gastrointestinal symptoms where extraintestinal manifestations predominate—for example, malignant diseases,3 osteopenia and bone fractures,4 infertility and unfavourable outcome of pregnancy,5,6 liver disorders and diseases,7 central nervous system involvement,8,9 autoimmune myocarditis,10 and potentially even the general development of autoimmune diseases.11 The classical extraintestinal disease is dermatitis herpetiformis where ingestion of gluten induces a blistering skin disease which can develop even in individuals with normal small intestinal morphology.12 Coeliac type gluten sensitivity is not restricted to villous atrophy, which in fact develops gradually from a normal mucosal morphology.13 Both gastrointestinal and extraintestinal symptoms, and even complications, may occur in the early stages when the mucosal morphology is still considered normal.8,14,15
The pathogenetic mechanisms of gluten induced small intestinal mucosal lesions are not fully understood,2 even less so in the case of extraintestinal manifestations. A general and disease specific phenomenon is the occurrence of circulating gluten dependent autoantibodies which target transglutaminase 2 (TG2).16,17 TG2 is also the antigen for the in vitro binding of coeliac IgA antibodies to intestinal and extraintestinal normal tissues, such as the liver, kidney, appendix, oesophagus, and umbilical cord sections, used in endomysial, reticulin, and jejunal antibody laboratory assays.18,19 These antibodies are present in the jejunal juice20 and are produced solely in the intestinal mucosa.21
Recent studies indicate that coeliac antibodies reduce the enzymatic activity of TG2 in vitro and,22 similar to monoclonal TG2 antibodies, inhibited transforming growth factor β (TGF-β) mediated epithelial cell differentiation in an in vitro small intestinal crypt villus axis model.23 Although these results prove that autoantibodies exert biological effects, their role in the immunopathology of the coeliac mucosal lesion, which is currently considered to be primarily a T lymphocyte mediated disorder, is a matter of debate. Indeed, it is not known to what extent TG2 autoantibodies reach their intracellularly produced enzyme autoantigen throughout the body in vivo, and whether autoantibody targeting precedes mucosal deterioration.
It has been repeatedly observed that the small intestinal epithelial basement membrane region contains deposited IgA in patients with active coeliac disease,24–26 and such IgA deposits have also been found in the subepithelial area and on subepithelial fibroblasts by immunoelectron microscopic studies.27 Whether this IgA is bound to any specific target is not known.
In this study, we hypothesised that in vivo deposited IgA targets TG2 and sought to establish whether extraintestinal TG2 specific autoantibody deposition occurs in coeliac patients with extraintestinal manifestations. Furthermore, we investigated if small intestinal subepithelial immunoglobulin deposition occurs in the early stages of disease development when the mucosal morphology is still normal.
MATERIALS AND METHODS
Jejunal biopsy samples
Samples from three sources were investigated. (i) Ten patients (aged 4.4–32 years at presentation, median 7.8) with developing coeliac disease and an initially normal small intestinal villous structure were studied. These patients were prospectively followed up, they consumed normal gluten containing food, and 4–33 months later (mean 9.3) developed overt coeliac disease with a flat mucosa. Initial samples were evaluated at the time of the first biopsy when the final diagnosis was not known. (ii) Eleven patients (aged 3.6–57 years, median 10.4) with untreated dermatitis herpetiformis were studied who had typical IgA deposits in the dermal papillae but normal jejunal mucosal architecture at presentation. (iii) The third group comprised 12 non-coeliac control patients (aged 1–17 years, median 4.6) with a normal small intestinal mucosa. Controls were negative for serum endomysial antibodies while all 10 patients in group (i) and five in group (ii) had circulating endomysial antibodies in serum. Clinical characteristics are presented in table 1 ▶.
Table 1.
Presenting clinical characteristics and symptoms in patients with a normal small intestinal villous architecture
| Patients with developing coeliac disease (n = 10) or dermatitis herpetiformis (n = 11) | Non-coeliac controls (n = 12) | |
| Diarrhoea | 5 | 6 |
| Short stature | 0 | 4 |
| Iron deficiency anaemia | 1 | 2 |
| Constipation | 1 | 0 |
| Asymptomatic family members of known coeliac patients | 3 | 0 |
| Rash | 11 | 0 |
Biopsies were obtained from the jejunum by Watson capsule and were cut into two pieces. One piece was fixed in formaldehyde for routine histology and the other was snap frozen in liquid nitrogen and stored at −70°C until use.
Other organ samples
A study was made of frozen tissue samples from six additional patients who underwent biopsies from organs other than the small bowel while they had active coeliac disease. All of these biopsies had been performed based on clinical indications independent of the evaluation of coeliac disease (table 2 ▶). Samples were traced retrospectively and included two lymph nodes, one skeletal muscle, two liver biopsy samples, and a non-inflamed appendix. As controls, one lymph node, three liver, and six appendix samples from non-coeliac patients were included. These had been removed in unrelated surgical interventions and were histologically normal. All biopsies were performed for diagnostic purposes and informed consent was obtained.
Table 2.
Clinical and laboratory characteristics in coeliac patients who had extrajejunal tissue removed
| Patient No | Sex/age (y) | Symptoms and signs | Removed tissue | Indication for biopsy/removal | Routine histology | Serum EMA titre | Jejunal biopsy histology | Outcome on a gluten free diet | Colocalisation of antibody deposits with TG2 |
| 1 | M/13 | Anaemia, iron deficiency, mesenterial lymph node and right testis enlargement | Mesenterial lymph node | Suspicion of malignancy | Normal* | 640 (IgA) | SVA | Marked clinical improvement, normal villi | Yes |
| Testis biopsy | Suspicion of malignancy | Normal | ND | ||||||
| 2 | F/14 | Diarrhoea, weight loss, anaemia, mesenterial lymph node enlargement | Mesenterial lymph node | Suspicion of malignancy | Normal | 640 (IgA) | SVA | Symptom free, refused a control jejunal biopsy | Yes |
| Appendix | Technical at laparoscopy | Normal | Yes | ||||||
| 3 | M/18 | Progressive gait disturbance, known but not treated coeliac disease | Skeletal muscle biopsy | Suspicion of progressive neuromuscular disease | Normal† | 40 (IgA) | PVA | Improved gait, normal villi | Yes |
| 4 | F/3 | Cystic fibrosis, anaemia, steatorrhoea, parvovirus B19 infection, elevated liver transaminase values | Liver biopsy | Acute liver enlargement | Steatosis | 320 (IgA) | SVA | Died of progressive liver failure. Near normal villi at autopsy | Yes |
| 5 | F/12 | Elevated liver transaminase values, selective IgA deficiency | Liver biopsy | HBsAg positivity | Normal | 5120 (IgG) | SVA | Symptom free, control jejunal biopsy not done | Yes |
| 6 | M/3 | Proteinuria, selective IgA deficiency | Kidney biopsy | Proteinuria | Mesangial IgG deposition | 1280 (IgG) | SVA | Symptom free, normal villi | ND |
*Also with immunohistochemistry for T and B cell markers.
†Also with histochemistry and electron microscopy.
EMA, serum endomysial antibodies; PVA, partial villous atrophy with crypt hyperplasia; SVA, subtotal villous atrophy; TG2, transglutaminase type 2; HBsAg, hepatitis B surface antigen; ND, not done.
Immunofluorescent studies
Unfixed 5 μm thick sections of patient tissues were investigated for deposited immunoglobulins by direct immunofluorescence using fluorescein isothiocyanate labelled rabbit antibodies against human IgA, IgG, and IgM (Dako AS, Glostrup, Denmark) at a dilution of 1:40 in phosphate buffered saline (PBS), pH 7.4. These stainings were also performed as multicolour procedures where patient immunoglobulins were shown in green and extracellular matrix proteins in red using rhodamine conjugated antimouse or antirabbit secondary antibodies, as described elsewhere.18,19 The primary antibodies used were monoclonal mouse antibodies against TG2 (CUB7402; NeoMarkers, Fremont, California, USA), and rabbit antibodies against fibronectin (Dako) and laminin (Dako), respectively, diluted 1:200 in PBS. For investigation of the relation of patient immunoglobulins to blood vessels, green was used for detection of patient IgA, red for TG2, and blue for an endothelial marker (rabbit antibodies against von Willebrand factor (Dako) followed by AMCA conjugated secondary antirabbit antibodies (Vector Laboratories, Burlingame, California, USA)).
Elution of IgA and TG2 from tissue sections
Unfixed cryosections were washed twice in PBS to remove blood and related antibodies. A solution of 0.25% chloroacetic acid (Fluka Chemie AG, Buchs, Switzerland) with 0.2 M/l NaCl, pH 2.7, was then used as the eluent28 and applied to the sections for 20 minutes. The eluates with chloroacetic acid were neutralised with 1 M/l imidazole, washed in PBS, concentrated by centrifugation on 10K membranes (Microsep, Pall Corporation, Ann Harbor, Michigan, USA), and lyophilised. Treated sections were washed twice in PBS and examined by immunofluorescence for the remaining IgA and TG2 in tissues, as described above.
Sections from extraintestinal samples, from six coeliac and three dermatitis herpetiformis jejunum samples, were investigated by immunofluorescence after choroacetic acid treatment. Lyophilised tissue eluates were prepared from a coeliac liver, lymph node, and a non-coeliac liver.
Detection of coeliac antibodies in serum samples and tissue eluates
Endomysial antibodies were investigated in serum samples and tissue eluates by an indirect immunofluorescent method on monkey oesophagus, normal human umbilical cord, jejunum, and appendix sections, as described elsewhere.18 The initial dilution was 1:2.5 in PBS, and samples non-reactive at this dilution were considered negative. Eluates were also tested on TG2 knockout mouse (TG2−/−) jejunum as a substrate, and on TG2−/− jejunum coated with human recombinant TG2 expressed in Escherichia coli,29 as previously described.19
Tissue eluates were tested for the presence TG2 and IgA by western blot30 and for TG2 specific IgA by ELISA using microtitre plates coated with fibronectin (Sigma, St Louis, Missouri, USA) and recombinant TG2, as described by Achyuthan and colleagues.31 Also, plates coated with rabbit antibodies against mouse IgG1 (ICN, Aurora; dissolved in 0.03 M bicarbonate buffer, pH 9.6) and CUB7402 monoclonal TG2 specific capture antibodies were applied to further verify the presence of both TG2 specific IgA and TG2 in the eluates. Bound IgA was detected with peroxidase conjugated rabbit antibodies against human IgA, as described previously.30
RESULTS
In vivo deposited IgA in the coeliac jejunum
In non-coeliac jejunal biopsy samples, IgA was detected only inside plasma cells and epithelial cells (fig 1A ▶). In contrast, clear subepithelial IgA positivity was found along the villous and crypt basement membranes in jejunal samples from all 10 coeliac patients, both in the developing stage of the disease when the villous architecture was still normal and also later when the mucosa had deteriorated to the typical flat lesion (fig 1B, C ▶, same patient).
Figure 1.
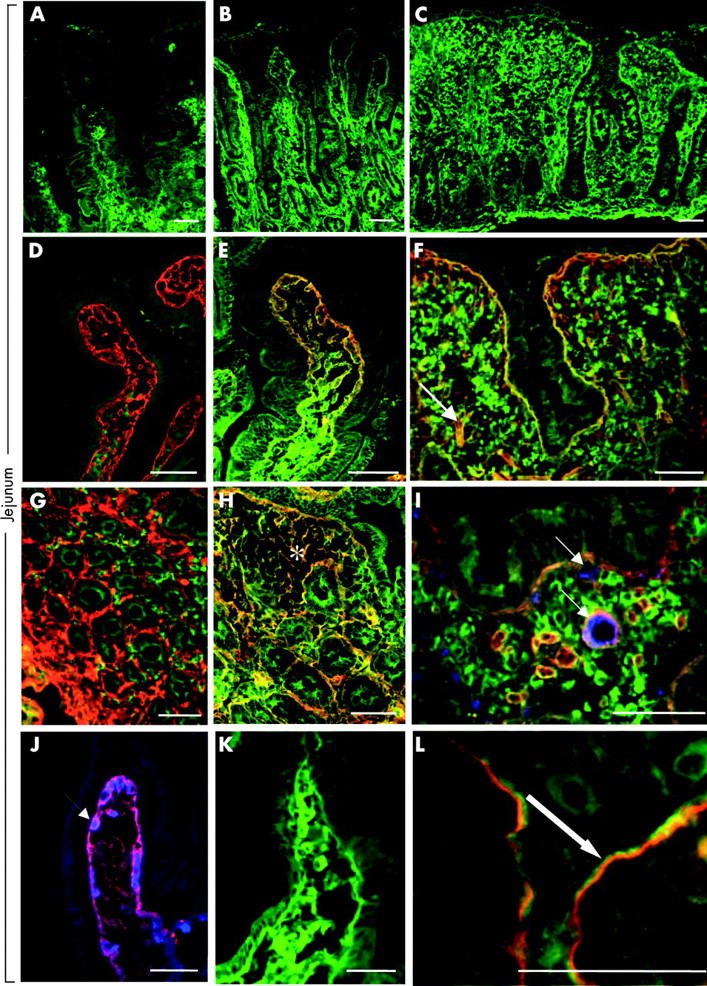
IgA (green) and transglutaminase 2 (TG2, red) in the normal jejunum (A, D, G), in the early developing stage of coeliac disease (B, E, H) and in the coeliac flat lesion (C, F, I). (B) Subepithelial IgA deposits in the villi and around crypts in the mucosa with a normal architecture and (C) in the flat mucosa developed four months later in the same patient. IgA deposits colocalised with TG2 (E, F, H. I), as indicated by the yellow merging of the labels. Both deposited IgA and TG2 were closely related to vessels shown in blue (I, J, arrows). (K) Similar jejunal IgA deposits in a patient with dermatitis herpetiformis and negative serum endomysial antibodies. (L) Crypt basement membrane laminin (red band) with coeliac IgA (green) deposits. There was only partial overlap (yellow) on the extracellular surface of the laminin. Bar = 50 μm.
This IgA positivity pattern was identical to the normal tissue localisation of TG2 (fig 1D, G ▶; red) which appeared merged into yellow in the coeliac mucosa due to colocalisation with IgA deposits labelled in green (fig 1E, F, H, I ▶). This colocalisation was also seen on reticulin fibres of lymphoid follicles (fig 1H ▶, asterisk) and on trabecular structures which appeared fragmented in the flat lesion (fig 1F ▶, arrow). IgA in plasma cells or inside crypt epithelial cells did not merge into yellow in either normal or coeliac mucosa. Both coeliac IgA and TG2 were arranged around mucosal vessels, shown in blue (fig 1 I, J ▶, arrows). Coeliac IgA was located on the extracellular surface of laminin (fig 1L ▶), and also showed a close relation to fibronectin (not shown). Similar IgA deposits were present in seven of 11 patients with dermatitis herpetiformis who had a normal jejunal villous structure at diagnosis. Two dermatitis herpetiformis patients who had clear jejunal IgA deposits were negative for circulating IgA class serum endomysial antibodies (fig 1K ▶).
Jejunal IgM and IgG positivity did not differ between coeliac and control samples.
In vivo deposited IgA in extrajejunal tissues
To establish whether coeliac IgA can reach TG2 in extraintestinal tissues, we examined by double immunofluorescence two coeliac lymph nodes, two liver samples, one appendix, and one skeletal muscle specimen from coeliac patients (table 2 ▶, figs 2 ▶, 3 ▶). Specimens were normal (cases 1, 2, 3, 5, and 6 in table 2 ▶) or showed only non-specific changes (case 4, liver steatosis; table 2 ▶) by conventional microscopy. All of these tissues contained abundant TG2 in endomysial localisations or around reticulin fibres both in non-coeliac controls (fig 2A ▶, 3B ▶) and in coeliacs (fig 2G, J ▶; fig 3D, F, H ▶, red). Tissue IgA was not related to endomysial or reticulin structures in non-coeliac lymph node (n = 1), appendix (n = 6), or liver (n = 3) samples and appeared only inside epithelial cells or in the centre of lymphoid follicles (fig 2A, D ▶; fig 3A, B ▶, green). However, in coeliac samples there were heavy IgA deposits on reticulin fibres also (lymph nodes, appendix, liver (fig 2B, E, H ▶; fig 3C ▶)) and on endomysial structures (muscular layer of the appendix, skeletal muscle (fig 2F, K ▶)) which were colocalised with tissue TG2 (fig 2C, I, L ▶; fig 3D ▶).
Figure 2.
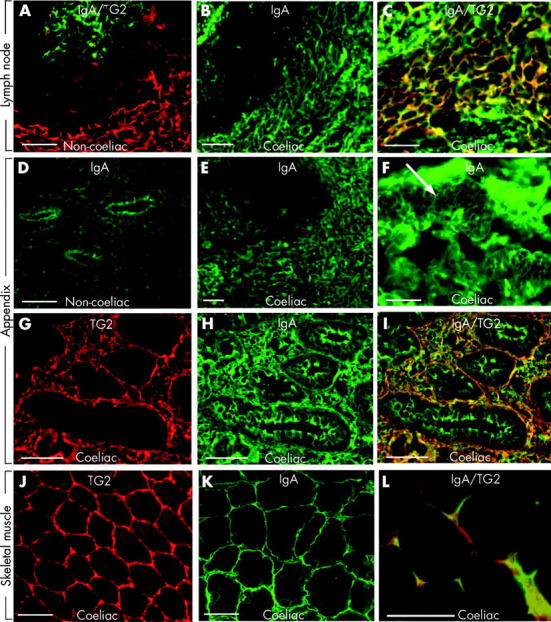
(A) Non-coeliac lymph node double stained for IgA (green) and transglutaminase 2 (TG2, red). (B) In vivo deposited IgA (green) on reticulin fibres in a lymph node from a coeliac patient (case 1 in table 2 ▶). (D) Normal appendix with IgA in epithelial cells. (E–I) Appendix from a coeliac patient (case 2) with in situ IgA deposits (green) on reticulin fibres (E), endomysium (F, arrow), and pericryptal subepithelial layer (H), corresponding to TG2 localisation in red (G). (J–L) Skeletal muscle from a coeliac patient (case 3) with an endomysial TG2 pattern (J) and endomysial IgA deposits (K, green). Colocalisation of IgA deposits with TG2 in all three coeliac specimens is indicated by yellow (C, I, L). There was no merging for IgA in the germinal centres or epithelial IgA (green in C and I). Bar = 50 μm.
Figure 3.
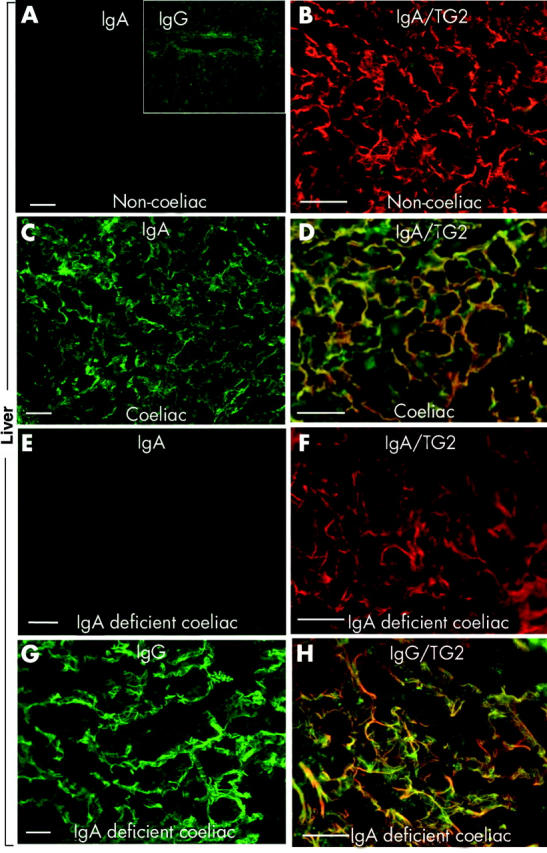
IgA or IgG (green) and transglutaminase 2 (TG2, red) in the liver of a non-coeliac patient (A–B), and from an IgA competent (C–D, case 4 in table 2 ▶) and IgA deficient (E–H, case 5) coeliac patient. In coeliac patients, there was IgA (in IgA deficiency IgG) deposition on reticulin fibres (C, G). Yellow in (D) and (H) indicates colocalisation of deposits with TG2. Bar = 50 μm.
In vivo deposited IgG in coeliac disease with IgA deficiency
In the case of IgA deficiency (cases 5 and 6 in table 2 ▶), IgA deposits were missing (fig 3E, F ▶) but TG2 related IgG deposits were seen in the liver (fig 3G, H ▶) and also on archive electron microscopic kidney pictures of case 6 (not shown).
Investigation of in vivo IgA-TG2 autoantigen binding
Colocalisation of in vivo deposited antibodies with TG2, both in the jejunum and also in extraintestinal tissues, strongly suggested that the specific target for coeliac antibody binding is TG2. To obtain direct evidence, we eluted deposited IgA from extraintestinal tissues for further studies.
We used chloroacetic acid to disrupt TG2 binding to fibronectin28 and to release the in vivo deposited IgA together with released TG2. Treatment of the sections resulted in disappearance of extracellular IgA deposits in sections from both the extrajejunal and jejunal coeliac samples, while IgA in plasma cells and epithelial cells remained visible on immunofluorescence (fig 4A, B ▶). Likewise, in tissues, TG2 became undetectable by monoclonal TG2 specific autoantibodies after elutions with choroacetic acid.
Figure 4.

Coeliac liver (A) and coeliac lymph node (B) double stained for IgA (green) and transglutaminase 2 (TG2, red) after elution with choroacetic acid which releases tissue TG2. (C) Demonstration of TG2 in the eluates by western blotting. (D) TG2 specific IgA detected in the eluates by ELISA using human recombinant TG2 antigen (bars 1–3) or anti-TG2 capture antibody (bars 4, 5): 1, 4: IgA eluted from a normal liver; 2: IgA eluted from the coeliac lymph node; 3, 5: IgA eluted from the coeliac liver. (E–K) Indirect immunofluorescent studies with the eluates. (E) IgA (green) eluted from coeliac liver bound to recombinant human TG2 coated on TG2 knockout mouse jejunum and merged into yellow (F) when TG2 was labelled in red, while IgA eluted from a normal liver did not bind (G–H). (I) Endomysial binding of IgA eluted from the coeliac lymph node to human umbilical cord. Binding of IgA eluted from the coeliac liver to monkey oesophagus endomysium (J) and human appendix subepithelial band (K). Bar = 50 μm.
TG2 specific IgA in coeliac tissue eluates
To ascertain that both TG2 and IgA are found in eluates, western blot and ELISA studies were conducted with concentrated eluates prepared from a coeliac and a non-coeliac control liver, and from a coeliac lymph node. A clear band at 80 kDa, corresponding to the molecular weight of TG2 was, as expected, equally detectable with monoclonal TG2 antibodies both in coeliac and non-coeliac eluates (fig 4C ▶). Human IgA was also detected both in coeliac and non-coeliac eluates (data not shown).
However, when the eluates were tested by ELISA for TG2 specific IgA class antibodies, high optical density values were obtained only with IgA eluted from the coeliac liver and lymph node (0.578 and 2.551, respectively) but not with IgA eluted from the control liver (0.052, representative values from two independent elution experiments). This finding indicates that IgA originating from non-coeliac tissues was not directed against TG2. The result was confirmed in an additional experiment where the eluates were added to ELISA plates coated with TG2 specific monoclonal antibodies and IgA was measured from the bound TG2 human IgA complexes; coeliac liver eluate gave an optical density of 0.493 versus 0.107 obtained with the control liver eluate (fig 4D ▶).
Similarly, IgA eluted from coeliac tissues bound to human recombinant TG2 applied on TG2−/− mouse tissues (fig 4E, F ▶), and also to the endomysial and reticulin structures of human umbilical cord (fig 4I ▶), monkey oesophagus (fig 4J ▶), and normal appendix (fig 4K ▶) sections. In contrast, IgA eluted from the non-coeliac liver did not bind to any of these structures (fig 4G, H ▶). Furthermore, there was no IgA binding to the TG2−/− mouse tissues with either the coeliac or non-coeliac eluates.
The experiments above collectively demonstrate that deposited IgA seen in coeliac tissues is targeted against TG2.
DISCUSSION
In the present study, we showed for the first time that in vivo targeting of TG2 in coeliac disease occurs in the form of in situ endomysial, reticulin, and jejunal subepithelial autoantibody binding. Patient IgA found deposited on extracellular TG2 in the liver, lymph nodes, and muscles indicates that the coeliac disease autoantigen is widely accessible to the intestinally produced21 circulating coeliac autoantibodies throughout the body. Based on clinical evidence of the occurrence of extraintestinal manifestations, coeliac disease should be regarded as a systemic disease and not solely involving the intestinal tract. The present primary observations may constitute one important denominator for gluten induced extraintestinal disease manifestations.
This study also showed that coeliac autoantibodies are deposited in the morphologically normal small intestinal mucosa before they can be detected in the circulation, and furthermore that the TG2 specific IgA deposition precedes the formation of the gluten induced flat jejunal lesion. This raises the possibility that autoantibodies against TG2, apart from T lymphocytes, contribute to the pathogenesis of mucosal atrophy in both coeliac disease and dermatitis herpetiformis. Also, in all other organs studied, TG2 was targeted by autoantibodies, as shown by the TG2 specificity of the IgA co-eluted together with TG2 from coeliac tissues. At the same time, conventional elution agents (for example, urea) capable of removing serum endomysial antibodies coated on tissue sections,32 did not extract any specific IgA from the same tissues (data not shown), which would strongly argue against the possibility that the eluted IgA represents circulating (blood) antibodies.
However, in the gluten dependent skin disease of dermatitis herpetiformis, IgA further targets type 3 transglutaminase in the skin.33
The coeliac disease autoantigen, TG2, is a ubiquitous cellular protein which also functions on the cell membrane associated with integrins.34 In the extracellular space, TG2 is involved in the cell-extracellular matrix interactions and also in the remodelling and stabilisation of the extracellular matrix.35 As coeliac antibodies have been shown to influence the protein cross linking enzymatic activity of TG2,22 their presence may interfere with the normal performance of the enzyme in the turnover of the extracellular matrix and cytokines. It may also be hypothesised that binding of autoantibodies to TG2 also disturbs TG2 mediated cellular adhesion. It has been shown that subepithelial blistering leading to loss of surface epithelial cells occurs in the coeliac mucosa,36 coincident with findings indicating that the most pronounced IgA deposition is characteristically seen subepithelially.24,26 None the less, ulceration is not a feature of coeliac disease. Our study also showed that TG2 stands in intimate relation with mucosal blood vessels where deposition of coeliac IgA (fig 1I ▶) might influence their nutritive and architectural function. There is remodelling of vessels in the coeliac flat lesion, and angiogenesis is very likely important for mucosal regeneration on diet when a normal villous structure without scarring is achieved.1
Coeliac disease may also induce architectural changes in extraintestinal tissues—for example, dental enamel defects in permanent teeth or loss of Purkinje cells in the brain.8,37 Our patients with extraintestinal manifestations and IgA autoantibody deposition in their organs evinced no specific abnormalities on conventional histology (table 2 ▶). However, some inflammation was present and it is possible that, as in the case of the jejunum,13 more time is required to develop advanced lesions. The most general pathology, scarring, seems to be related to the bioactivity of TG235 and may be inhibited by the presence of antibodies. The observed clinical symptoms and signs (for example, size of the lymph nodes, muscular weakness, and proteinuria) were clearly responsive to gluten withdrawal, which suggests an ongoing pathology in the respective organs. In situ endomysial antibody deposition would also be a reasonable case for the gastrointestinal motility disorder observed in other clinical studies.38 Direct evaluation of the effect and dynamics of extraintestinal TG2 antibody deposition is especially difficult as repeated biopsies were not possible in our cases. In addition, our liver patients also had independent hepatic diseases (case 4: cystic fibrosis, parvovirus B19 infection39; case 5: hepatitis B infection). TG2 specific autoantibodies found in the morphologically normal appendix again indicate that a clinically silent deposition of TG2 specific antibodies in various organs can occur in any coeliac patient. It remains to be established which factors determine organ manifestations or whether there are contributing factors directing the antibodies to particular organs.
The ability of coeliac autoantibodies to inhibit the bioactivity of TG222 and the resulting decrease in the availability of active TGF-β23 could also be part of the mechanisms which elicit the diverse extraintestinal disease entities. In fact, TGF-β is known as an elementary regulatory growth factor in organogenesis and tissue homeostasis maintenance, in addition to its known function as a proinflammatory and immunosuppressive factor. Furthermore, as the present results show the self-antigen to be targeted by immunoglobulins early on in disease development, it is intriguing to hypothesise that an initially ongoing Th2-type mucosal inflammation passes on to the classical Th1-type inflammatory process only later, as the mucosa deteriorates.
As the histological changes in coeliac organ manifestations are non-specific, examination of TG2 specific immunoglobulin deposition by direct immunofluorescence might be a useful diagnostic tool in the differential diagnosis of organ diseases of unknown aetiology. For example, lymph node enlargement, hitherto not acknowledged as a presenting symptom in coeliac disease, was a prominent feature in two of our patients.
TG2 related IgA deposits in the morphologically normal jejunum were predictive of forthcoming overt coeliac disease with villous atrophy. They may thus be regarded as a further marker of developing coeliac disease and coeliac disease latency, and a promising diagnostic tool in cases with low grade enteropathy. In agreement with previous studies,24,26 our results support the notion that subepithelial IgA deposits are specific for gluten induced enteropathy. However, further studies are needed to confirm this in larger patient groups and to explore the kinetics of IgA deposition in relation to gluten intake and intraepithelial lymphocyte counts. Moreover, it should be established whether humoral targeting of TG2 occurs in gluten induced peripheral or central nervous system diseases, or heart, bone, or reproductive system involvement.
In conclusion, the widespread in situ endomysial, reticulin, and jejunal binding of TG2 specific IgA antibodies found in the tissues of our coeliac patients with developing disease or extraintestinal organ manifestations strongly supports the concept that humoral immunity is involved in pathogenesis of coeliac disease.
Acknowledgments
The authors thank Mária Géczy, Margit Lörincz, Anikó Marosi, Erika Molnár, Anikó Nagy, and Rita Újhelyi for collaboration in the clinical care of the patients, Zoltán Szentirmai, National Institute of Oncology, Budapest, Hungary, for kindly providing frozen sections from one of the coeliac lymph nodes, and Gerry Melino, University of Rome “Tor Vergata”, Italy, for the generous donation of TG2 knockout mice. The study was supported by the Medical Research Fund of Tampere University Hospital, the Päivikki and Sakari Sohlberg Foundation, the Foundation of the Friends of the University Children’s Hospitals in Finland, and the Finnish Coeliac Society.
Abbreviations
ELISA, enzyme linked immunosorbent assay
PBS, phosphate buffered saline
TG2, transglutaminase type 2 or tissue transglutaminase
TGF-β, transforming growth factor β
REFERENCES
- 1.Mäki M, Collin P. Coeliac disease. Lancet 1997;349:1755–9. [DOI] [PubMed] [Google Scholar]
- 2.Schuppan D. Current concepts of celiac disease pathogenesis. Gastroenterology 2000;119:234–42. [DOI] [PubMed] [Google Scholar]
- 3.Holmes GK. Coeliac disease and malignancy. Dig Liver Dis 2002;34:229–37. [DOI] [PubMed] [Google Scholar]
- 4.Bernstein CN, Leslie WD, Leboff MS. AGA technical review on osteoporosis in gastrointestinal diseases. Gastroenterology 2003;124:795–841. [DOI] [PubMed] [Google Scholar]
- 5.Sher KS, Mayberry JF. Female fertility, obstetric and gynaecological history in coeliac disease: a case control study. Acta Paediatr Suppl 1996;412:76–7. [DOI] [PubMed] [Google Scholar]
- 6.Collin P, Vilska S, Heinonen PK, et al. Infertility and coeliac disease. Gut 1996;39:382–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaukinen K, Halme L, Collin P, et al. Celiac disease in patients with severe liver disease: gluten-free diet may reverse hepatic failure. Gastroenterology 2002;122:881–8. [DOI] [PubMed] [Google Scholar]
- 8.Hadjivassiliou M, Gibson A, Davies-Jones GA, et al. Does cryptic gluten sensitivity play a part in neurological illness? Lancet 1996;347:369–71. [DOI] [PubMed] [Google Scholar]
- 9.Kieslich M, Errazuriz G, Posselt HG, et al. Brain white-matter lesions in celiac disease: a prospective study of 75 diet-treated patients. Pediatrics 2001;108:E21. [DOI] [PubMed] [Google Scholar]
- 10.Frustaci A, Cuoco L, Chimenti C, et al. Celiac disease associated with autoimmune myocarditis. Circulation 2002;105:2611–18. [DOI] [PubMed] [Google Scholar]
- 11.Ventura A, Magazzu G, Greco L. Duration of exposure to gluten and risk for autoimmune disorders in patients with celiac disease. Gastroenterology 1999;117:297–303. [DOI] [PubMed] [Google Scholar]
- 12.Fry L. Dermatitis herpetiformis. Baillière Clin Gastroenterol 1995;9:371–93. [DOI] [PubMed] [Google Scholar]
- 13.Troncone R. Latent coeliac disease in Italy. Acta Paediatr 1995;84:1252–7. [DOI] [PubMed] [Google Scholar]
- 14.Freeman HJ, Chiu BK. Multifocal small bowel lymphoma and latent celiac sprue. Gastroenterology 1986;90:1992–7. [DOI] [PubMed] [Google Scholar]
- 15.Kaukinen K, Mäki M, Partanen J, et al. Celiac disease without villous atrophy: revision of criteria called for. Dig Dis Sci 2001;46:879–87. [DOI] [PubMed] [Google Scholar]
- 16.Mäki M. The humoral immune system in coeliac disease. Bailliere Clin Gastroenterol 1995;9:231–49. [DOI] [PubMed] [Google Scholar]
- 17.Dieterich W, Ehnis T, Bauer M, et al. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nature Med 1997;3:797–801. [DOI] [PubMed] [Google Scholar]
- 18.Korponay-Szabó IR, Sulkanen S, Halttunen T, et al. Tissue transglutaminase is the target in both rodent and primate tissues for celiac disease-specific autoantibodies. J Pediatr Gastroenterol Nutr 2000;31:520–7. [DOI] [PubMed] [Google Scholar]
- 19.Korponay-Szabó IR, Laurila K, Szondy Z, et al. Missing endomysial and reticulin binding of coeliac antibodies in transglutaminase 2 knockout tissues. Gut 2003;52:199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mawhinney H, Lowe AHG. Anti-reticulin antibody in jejunal juice in coeliac disease. Clin Exp Immunol 1975;21:394–8. [PMC free article] [PubMed] [Google Scholar]
- 21.Marzari R, Sblattero D, Florian F, et al. Molecular dissection of the tissue transglutaminase autoantibody response in celiac disease. J Immunol 2001;166:4170–6. [DOI] [PubMed] [Google Scholar]
- 22.Esposito C, Paparo F, Caputo I, et al. Anti-tissue transglutaminase antibodies from coeliac patients inhibit transglutaminase activity both in vitro and in situ. Gut 2002;51:177–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Halttunen T, Mäki M. Serum immunoglobulin A from patients with celiac disease inhibits human T84 intestinal crypt epithelial cell differentiation. Gastroenterology 1999;116:566–72. [DOI] [PubMed] [Google Scholar]
- 24.Shiner M, Ballard J. Antigen-antibody reactions in jejunal mucosa in childhood celiac disease after gluten challenge. Lancet 1972;1:1202–5. [DOI] [PubMed] [Google Scholar]
- 25.Lancaster-Smith M, Packer S, Kumar PJ, et al. Immunological phenomena in the jejunum and serum after reintroduction of dietary gluten in children with treated coeliac disease. J Clin Pathol 1976;29:592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kárpáti S, Kósnai I, Török E, et al. Immunoglobulin A deposition in jejunal mucosa of children with dermatitis herpetiformis. J Invest Dermatol 1988;91:336–9. [DOI] [PubMed] [Google Scholar]
- 27.Jos J, Labbe F. Ultrastructural localisation of IgA globulins in normal and coeliac intestinal mucosa using immunoenzymatic methods. Biomedicine 1976;24:425–34. [PubMed] [Google Scholar]
- 28.Radek JT, Jeong JM, Murthy SN, et al. Affinity of human erythrocyte transglutaminase for a 42-kDa gelatin-binding fragment of human plasma fibronectin. Proc Natl Acad Sci U S A 1993;90:3152–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ambrus A. Bányai I. Weiss MS, et al. Calcium binding of transglutaminases: A 43Ca NMR study combined with surface polarity analysis, J Biomol Struct Dyn 2001;19:59–74. [DOI] [PubMed] [Google Scholar]
- 30.Sulkanen S, Halttunen T, Laurila K, et al. Tissue transglutaminase autoantibody enzyme-linked immunosorbent assay in detecting celiac disease. Gastroenterology 1998;115:1322–8. [DOI] [PubMed] [Google Scholar]
- 31.Achyuthan KE, Goodell RJ, Kennedye JR, et al. Immunochemical analyses of human plasma fibronectin-cytosolic transglutaminase interactions. J Immunol Methods 1995;180:69–79. [DOI] [PubMed] [Google Scholar]
- 32.Lock RJ, Gilmour JE, Unsworth DJ. Anti-tissue transglutaminase, anti-endomysium and anti-R1-reticulin autoantibodies—the antibody trinity of coeliac disease. Clin Exp Immunol 1999;116:258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sárdy M, Kárpáti S, Merkl B, et al. Epidermal transglutaminase (TGase 3) is the autoantigen of dermatitis herpetiformis. J Exp Med 2002;195:747–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akimov SS, Krylov D, Fleischman LF, et al. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol 2000;148:825–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffin M, Casadio R, Bergamini CM. Transglutaminases: nature’s biological glues. Biochem J 2002;368:377–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kainulainen H, Rantala I, Collin P, et al. Blisters in the small intestinal mucosa of coeliac patients contain T cells positive for cyclooxygenase 2. Gut 2002;50:84–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aine L. Coeliac-type permanent-tooth enamel defects. Ann Med 1996;28:9–12. [DOI] [PubMed] [Google Scholar]
- 38.Cucchiara S, Bassotti G, Castellucci G, et al. Upper gastrointestinal motor abnormalities in children with active celiac disease. J Pediatr Gastroenterol Nutr 1995;21:435–42. [DOI] [PubMed] [Google Scholar]
- 39.Granot E, Miskin H, Aker M. Monoclonal anti-CD52 antibodies: A potential mode of therapy for Parvovirus B19 hepatitis. Transplant Proc 2001;33:2151–3. [DOI] [PubMed] [Google Scholar]