

Abstract
Background and aim: Significant telomere shortening of hepatocytes is associated with replicative senescence and a non-dividing state in chronic liver disease, resulting in end stage liver failure and/or development of hepatocellular carcinoma. To prevent critical telomere shortening in hepatocytes, we have focused on oestrogen dependent transactivation of the human telomerase reverse transcriptase (hTERT) gene as a form of telomerase therapy in chronic liver disease.
Methods: We examined expression of hTERT mRNA and its protein, and telomerase activity (TA) in three human normal hepatic cell lines (Hc-cells, h-Nheps, and WRL-68) before and after treatment with 17β-oestradiol. The effects of exogenous oestradiol administration were examined in a carbon tetrachloride (CCl4) induced model of liver fibrosis in rats.
Results: Expression of hTERT mRNA and its protein was upregulated by oestradiol treatment. Telomere length decreased in Hc-cells and h-Nheps with accumulated passages whereas with long term oestradiol exposure it was greater than without oestradiol. The incidence of β-galactosidase positive cells, indicating a state of senescence, decreased significantly in oestradiol treated cells in comparison with non-treated cells (p<0.05). TA in both male and female rats with CCl4 induced liver fibrosis was significantly higher with oestradiol administration than without (p<0.05). Long term oestradiol administration markedly rescued the hepatic telomere from extensive shortening in both male and female rats.
Conclusion: These results suggest that oestradiol acts as a positive modulator of the hTERT gene in the liver. Oestrogen dependent transactivation of the hTERT gene is a new strategy for slowing the progression of chronic liver disease.
Keywords: telomerase, telomerase reverse transcriptase, chronic liver disease, oestrogen receptor, telomeric repeat elongation assay
Progression of chronic liver disease that culminates in cirrhosis is characterised by chronic hepatocyte death and renewal over a period of years. In the process, abnormal patterns of hepatocyte regeneration and fibrous scarring are promoted, and these architectural abnormalities accelerate to compromise hepatocyte function, causing systemic life threatening complications. The liver tries to compensate for the dysfunction by producing new hepatocytes. However, the hepatocytes eventually stop cell division, and this in turn results in end stage organ failure. To attempt a new treatment strategy for slowing the progression of chronic liver disease, several researchers have extensively studied the mechanism of fibrotic or cirrhotic initiation and progression at the level of cellular and molecular events.1–22 Two major hypotheses have been proposed for the molecular mechanism governing the progressive induction of cirrhosis during its long latency.
One is that hepatic stellate cells (HSCs) are responsible for hepatofibrogenesis.15–22 With the stimulation of inflammation, toxification, and activation signals released from the destroyed hepatocytes themselves, HSCs are converted to myofibroblast-like cells.19–21 Activated HSCs secrete extracellular matrix components15,16 and fibrosis implementing factors, such as transforming growth factor β1 (TGF-β1)11–14 and platelet derived growth factor.17 These cellular and molecular mechanisms presumably contribute to initiation and/or progression of hepatic fibrosis.
As the second key aspect of terminal liver failure, hepatocyte proliferative arrest has been linked to several aetiological factors, including altered hepatocyte-matrix interactions, growth inhibition by abundant TGF-β1,11–14 and/or critical telomere shortening.1–6
The telomere is a special cap-like structure at the end of eukaryotic chromosomes, composed of a tandem repeat of TTAGGG.23 Continuous shortening of the telomere occurs during each cell division, due to an end replication problem. Significant telomere shortening is associated with replicative senescence and a non-dividing state, the “Hayflic limit/mortality stage 1 (M1)”,24 through activation of the pRb and p53 pathway.25 More continuous telomere shortening beyond the Hayflic limit leads to loss of telomere capping activity, culminating in severe chromosomal instability and massive cell death, referred to as “crisis/mortality stage 2 (M2)”. The telomere hypothesis2 proposes that chronic liver injury induces continuous cell death and regeneration, resulting in critical telomere shortening, which in turn culminates in hepatocyte replicative senescence or crisis (fig 1A ▶).
Figure 1.
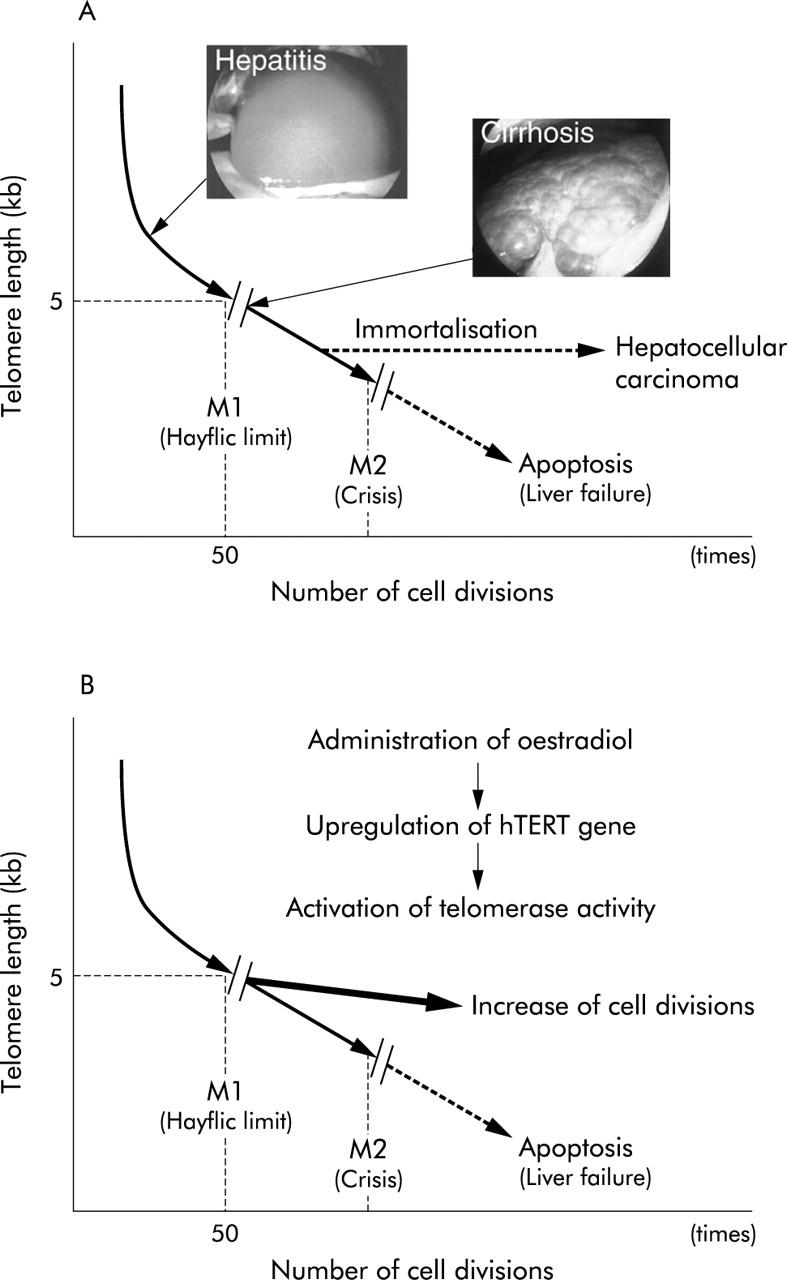
The telomere hypothesis (A) and the strategy of oestrogen dependent telomerase therapy (B) for chronic liver disease. (A) Chronic liver injury induces continuous cell death and regeneration resulting in critical telomere shortening, which in turn culminates in hepatocyte replicative senescence (M1) or crisis (M2). Critical telomere shortening produces severe chromosomal instability that may play an important role in hepatocyte immortalisation followed by the development of hepatocellular carcinoma. (B) Oestrogen dependent transactivation of the hTERT gene prevents extensive telomere shortening, thus prolonging the lifespan of hepatocytes in chronic liver disease.
Some researchers have discovered that hepatocytes in liver cirrhosis have shorter telomeres than those from age matched healthy hepatocytes.1–6 Moreover, quantitative results for telomere length were well correlated with senescence associated β-galactosidase staining, a biomarker for senescence, in hepatocytes of humans with cirrhosis.1 To test this hypothesis, Rudolph and colleagues18 studied telomerase deficient mice (mouse telomerase RNA component: mTERC−-/−). Telomere dysfunction in mice was associated with defects in liver regeneration and accelerated the development of liver cirrhosis in response to chronic liver injury. Telomerase gene delivery by adenovirus vectors prevented cirrhosis formation in the mice with short telomeres.18 These data strongly support the telomere hypothesis and encourage evaluation of telomerase therapy for chronic liver disease.
We have been focusing on the telomerase catalytic subunit (human telomerase reverse transcriptase: hTERT) as a form of telomerase therapy for chronic liver disease. Some indirect and direct evidence has been documented for a possible association between hormonal systems and telomere length.26–31 It is well known that administration of exogenous oestradiol suppresses the progression of hepatic fibrosis and decreases serum levels of hepatic transaminase.15,22 Oestradiol downregulates the release of extracellular matrix components and fibrosis implementing factors in HSCs. On the other hand, several studies have indicated that oestrogen dependent transactivation of the hTERT gene is carried out by direct interaction of activated oestrogen receptor (OR) with an imperfect oestrogen response element in the hTERT promoter.26 Exogenous oestradiol administration may not only help to suppress induction of hepatic fibrosis by HSCs15,22 but also prevent critical telomere shorting of hepatocytes by transactivation of the hTERT gene (fig 1B ▶). The present study examined the effects of oestradiol on telomerase activity (TA) and telomere length in human normal hepatic cultured cells and a carbon tetrachloride (CCl4) induced liver cirrhosis model in rats.
MATERIALS AND METHODS
Cell culture
One hepatocellular carcinoma cell line, Hep 3B (Cell Resource Centre for Biomedical Research, Tohoku University, Sendai, Japan) and three human normal hepatocyte cell lines, Hc-cells (Applied Cell Biology Research Institute, Human Hepatocyte Cell culture #3716, Kirkland, Washington, USA), h-Nheps (BioWhittaker, Walkersville, Maryland, USA), and WRL-68 (American Type Culture Collection, Rockville, Maryland, USA) were obtained commercially. They were maintained under the recommended conditions. For the oestradiol induction assay, they were cultured in phenol red-free medium (RPMI-1640; Invitrogen, Grand Island, New York, USA) supplemented with 10% charcoal/dextran treated fetal bovine serum (HyClone Laboratories Inc., Logan, Utah, USA).
For the evaluation of telomerase activity (TA), cells were seeded at 1×105 cells per 100 mm petri dish, and subsequently cultured in the above mentioned medium without oestradiol. After 24 hours of incubation, the medium was replaced with that containing 1, 10, or 100 nM 17β-oestradiol (oestradiol: 1.3.5(10)-estratriene-3, 17β-diol; Sigma, St Louis, Missouri, USA). Telomerase was extracted after 48 hours of exposure to oestradiol.
For determination of terminal restriction fragment (TRF) length, all cells were continuously exposed to oestradiol (10 nM) or untreated. We started a long term culture with continuous oestradiol exposure after three passages from the time when cells were purchased. Cells were passaged weekly at 75% confluence using 0.25% trypsin/0.02% ethylenediaminetetraacetic acid (Invitrogen) for detachment and seeding of new dishes using a split ratio of 1:10. DNAs of cells were extracted at the 10th, 20th, and 30th passages.
Preparation of animals
Forty Fisher rats—20 males and 20 females—(CLEA Japan Inc, Tokyo, Japan) at five weeks old, weighing 102 (13) g, were used. They were divided into two groups: CCl4 alone (group A) and CCl4+oestradiol (group B). Twenty rats—10 males and 10 females—were prepared as a non-treatment control. All rats were injected intramuscularly with 10% CCl4 in olive oil at a dose of 1 ml/kg body weight twice weekly. Rats in group B additionally received an intraperitoneal injection of oestradiol (1.3.5(10)-estratriene-3, 17β-diol 17-pentanoate; Mochida Pharmaceutical Co., Tokyo, Japan) in olive oil at a dose of 3.3 mg/kg body weight twice weekly. All animals were anaesthetised with sodium pentobarbital (40 mg/kg body weight intraperitoneally) and killed by terminal exsanguination from the inferior vena cava at 24 and 48 weeks. Japanese national guidelines for the care of animals were followed.
Western blot analysis
Cell lines and rat liver tissues were dissolved in cell lysis buffer (50 mM HEPES (pH 7.5)/1 mM EDTA/150 mM NaCl/2.5 mM EGTA/1.0% NP-40) and the clear supernatants were collected after a 30 minute rotation. Protein concentration in the lysates was determined spectrophotometrically using an RC DC Protein Assay Kit (Bio-Rad, Hercules, California, USA), and 5 μg of extract was subjected to sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis. Proteins were transferred to Hybond-P membranes (Amersham Biosciences, Buckinghamshire, UK). A primary polyclonal antibody against ORα (MC-20; Santa Cruz Biotechnology, Santa Cruz, California, USA; diluted 1:5000) or a monoclonal antihuman TERT antibody (NCL-hTERT; Novocastra Laboratories Ltd, Newcastle upon Tyne, UK) was applied to the membrane for 60 minutes at room temperature. The secondary antibody, antirabbit IgG or antimouse IgG (Amersham Biosciences) diluted 1:10 000 in blocking buffer, was applied for 45 minutes at room temperature. Both expressions were detected by an ECL (enhanced chemiluminescence) Western Blotting Detection System Kit (Amersham Biosciences).
Telomerase assays
TA was measured by the modified telomeric repeat elongation assay (TRE) assay, which is a rapid and sensitive direct telomerase assay32 that has many advantages over conventional polymerase chain reaction (PCR) based methods. The most significant advantage is that the TA of somatic cells or tissues can be evaluated quantitatively, eliminating inhibitors of DNA polymerase in the reaction mixture. The 5′-biotinylated oligomers (TS-5′B: biotin-5′AAT CCG TCG AGC AGA GTT AGG GTT AGG GTT AGG GTT AGG GTT AG –3′) were immobilised on streptavidin pretreated dextran sensor surfaces in situ using a Biacore apparatus (Biacore 3000; Biacore AB, Uppsala, Sweden). In the modified TRE assay, we also immobilised identical oligomers that were biotinylated at the 3′ end (TS-3′B) on a reference flow cell. The modification achieved more precise quantification of telomerase activity. Conditions and reaction buffers have been described previously.32 Briefly, cell extracts containing telomerase were extracted by the conventional method and diluted in TRE buffer (10 mM HEPES, pH 7.4/150 mM NaCl/10 mM MgCl2/2.5 mM dNTP/10 mM EGTA) to 0.1 mg/ml. They were applied at 5 μl/min across the chip surface for five minutes at 37°C. The chip surface was regenerated by washing with a 100 μl pulse of 1% SDS in 10 mM HEPES, which removed all protein, at a flow rate of 100 μl/min. After regeneration, the flow rate returned to 5 μl/min. We defined the elongation value (e value) as the difference in resonance unit (RU) between baseline levels before the extract injection and after the regeneration phase. All values were determined from 3–6 independent assays, and the coefficients of variation of all data were confirmed to be below 5%. The increased RU value following immobilisation was used to determine the moles of DNA on the chip surface, using an empirical relationship in which 1500 RU (1 RU = 1.2 pg/m2) was equivalent to 1.8 ng of DNA (1.276×10−13 mol of TS-5′B oligomer). An extension of one base on all TS-5′B oligomers would thus result in an increase of approximately 33.8 RU.
Five independent assays were performed on each telomerase sample extracted from hepatocyte cell lines. For analysis in the animal model of CCl4 induced liver fibrosis, we performed the TRE assay five times and calculated the mean e value for each rat.
Real time quantitative-PCR for hTERT mRNA
Total RNA was extracted with Triazol reagent (Invitrogen) and cDNA was synthesised using a Thermoscript reverse transcription-PCR kit (Invitrogen) and oligo-dT primer. We performed the real time quantitative-PCR (RQ-PCR) assay using an ABI Prism 7700 Sequence Detector (Applied Biosystems, Foster City, California, USA). Sequences of the primers and probe were as follows: hTERT-F, CAG CCT CCA GAC GGT GTG; hTERT-R, GCG ACA TCC CTG CGT TCT; and hTERT-P, FAM-CCT GCT GCA GGC GTA CAG GTT TCA C-TAMURA. The reaction mixture contained cDNA equivalent to 200 ng of total RNA, 200 nmol/l each primer, 5 μM fluorogenically labelled probes, and 25 μl of TaqMan Universal PCR Master Mix (Applied Biosystems) in a final volume of 50 μl. As the initial step, AmpErase uracil-N-glycosylase, which can be used to control carryover contamination, was activated for two minutes at 50°C, followed by 10 minutes at 95°C to activate AmpliTaq Gold DNA polymerase. DNA was subjected to 50 cycles of a two step PCR consisting of a 15 second denaturation step at 95°C and a one minute combined annealing/extension step at 60°C.
The assay specific standard curve was constructed by plotting the threshold cycle against the known copy number of each positive control plasmid. Control plasmids were made by the PCR products amplified from Hep 3B. The product was ligated to pGEM-T Easy Vectors (Promega, Madison, Wisconsin, USA) and transformed into DH5α competent cells (Toyobo, Tokyo, Japan). Plasmids were diluted in a precise series, ranging from 5 pg to 0.005 fg (2×106 to 2 copies). For normalisation, the copy number of GAPDH was used as an internal control.
Five independent assays were performed on each RNA sample extracted from hepatocyte cell lines. For analysis in the animal model of CCl4 induced liver fibrosis, we performed five independent assays, and calculated mean normalised copy number for each rat.
TRF length analysis
DNA was isolated and then digested with the restriction enzyme HinfI. DNA (10 μg/lane) was loaded onto a 0.8% agarose gel and electrophoresed for 12–20 hours at 60 V. The gel was denatured in 0.5 M NaCl and 0.5 M NaOH for 40 minutes, and neutralised in 1.5 M NaCl and 1 M Tris HCl (pH 7.5) for 40 minutes. Then, DNA was transferred onto Hybond-N membrane (Amersham Biosciences) in 20×SSC for two hours using a VacuGene-XL blotting apparatus (Amersham Biosciences). The membrane was hybridised to a 5′-[32P]-(TTAGGG)4 probe in Perfect Hyb plus hybridisation buffer (Sigma) at 42°C for 12 hours, then finally given two washes in 0.1% SDS, 1×SSC at 42°C, prior to exposure to Kodak XAR film (Kodak, Tokyo, Japan) at −80°C for 1–2 days. Each lane was scanned with a densitometer and mean TRF length was calculated.
Senescence associated β-galactosidase staining
Senescence associated β-galactosidase staining was performed as described previously.33 In brief, cells were fixed in 3% formaldehyde for five minutes, followed by three washes in phosphate buffered saline (PBS) at room temperature. They were immersed in freshly prepared senescence associated β-galactosidase staining solution (1 mg/ml 5-bromo-4-chloro-3-indolyl-D galactoside in DMF/40 mM citric acid/sodium phosphate (pH 6.0)/5 mM potassium ferrocyanide/5 mM potassium ferricyanide/150 mM NaCl/2 mM MgCl2) and incubated at 37°C for 14–16 hours. To detect lysosomal β-galactosidase, citric acid/sodium phosphate (pH 4.0) was applied. Two researchers microscopically evaluated 500 cells five times, and determined the percentage of those staining positive for β-galactosidase.
Quantitative assessment of hepatic fibrosis and hepatocyte proliferation
Sections (4 μm thick) were cut from formalin fixed paraffin-embedded samples of liver with CCl4 induced cirrhosis and stained with standard Masson’s trichrome. Using a Kontron Elektronik Imaging System KS 300 (Kontron Elektronik, München, Germany), fibrotic areas were measured. We acquired approximately 20 images in each 2×1.5 cm specimen from each rat. Severity of liver fibrosis was expressed as a percentage according to the formula: (fibrotic areas/fibrotic and hepatic areas) × 100.
For assessment of hepatocytes proliferation, we examined the Ki67 labelling index (MIB-1; Novocastra). Antigen was obtained by a microwave based antigen retrieval method with 10 mM citrate buffer (pH 6.0) for 15 minutes. After deparaffinisation and antigen retrieval, sections were washed three times in PBS (3×5 minutes) and immersed in 0.3% hydrogen peroxide in methanol for 30 minutes to block endogenous peroxidase activity. After three washes with PBS (3×5 minutes), sections were incubated with 5% bovine serum albumin for 10 minutes to block non-specific reactions. Then, sections were incubated with anti-Ki67 antibody (dilution 1:100) overnight at 4°C. Secondary antibody and peroxidase labelling were performed with a Simple Stain MAX-PO Kit (Nichirei, Tokyo, Japan), colourisation was produced by DAB substrate (Dako, Glostrup, Denmark), and counterstaining was performed with Mayer’s haematoxylin. We counted 1000 hepatocytes in total, and the Ki67 labelling index was expressed as the number of Ki67 positive hepatocytes per 1000 hepatocytes.
Statistical analysis
For comparison of the e value of rat liver tissues measured by TRE assay among controls, group A, and group B, analysis of variance (ANOVA) was performed followed by the least significant difference method. For comparison of the number of senescent cells between oestradiol and non-oestradiol treated cell lines, the Mann-Whitney U test was used. ANOVA was used for the remaining statistical analyses of differences between oestradiol treated and non-oestradiol treated cell lines and animals, using a significance level of p<0.05.
RESULTS
Expression of ORα and hTERT in human normal hepatic cell lines
All human normal hepatic cell lines (Hc-cells, h-Nheps, and WRL-68) expressed ORα to various degrees (fig 2A ▶). Overexpression of hTERT protein was observed in WRL-68 (fig 2B ▶). WRL-68 had been established from a fetal liver and is well known as a telomerase positive cell line. Hc-cells were weakly positive and h-Nheps was negative for hTERT protein (fig 2B ▶). Quantitative results for hTERT mRNA measured by RQ-PCR assay were compatible with the results of western blotting (table 1 ▶).
Figure 2.

Western blot analyses for oestrogen receptor α (ORα) (A) and hTERT (B) in three normal human hepatic cell lines. Cell lysate (5 μg) was loaded in each lane. Expression of ORα varied among cell lines (A). Overexpression of hTERT protein was observed in WRL-68, which is a known telomerase positive cell line, established from fetal liver (B).
Table 1.
Results of real time quantitative-polymerase chain reaction assay for measuring hTERT mRNA in human normal hepatic cell lines
| Cell line | Copy number of hTERT mRNA/103 GAPDH | |
| Oestradiol (−) | Oestradiol (+)† | |
| Hc-cells | 10.9 (4.23) | 115 (23.3)* |
| WRL-68 | 468 (42.0) | 757 (130)* |
| h-Nheps | 0 | 8.06 (4.44)* |
Values are means (SD) of five independent assays.
†Final concentration of oestradiol in the supernatant was 10 nM.
*p<0.05 by analysis of variance.
Oestradiol induction assay on hTERT expression and telomerase activity in human normal hepatic cell lines
In all human normal hepatic cell lines, a concentration dependent increase in hTERT protein expression by oestradiol induction was observed (fig 3 ▶) although this was weaker in h-Nheps than in the two other cell lines (Hc-cells and WRL-67) (fig 3C ▶). Quantitative results for hTERT mRNA measured by RQ-PCR assay were compatible with the results of western blotting (table 1 ▶). TAs of Hc-cells and h-Nheps were significantly increased after treatment with oestradiol (p<0.05) whereas that of WRL-68 was slightly increased. However, the difference was not significant (table 2 ▶, fig 4A, 4B ▶ ▶). Increased TAs of three normal hepatic cell lines on oestradiol induction, ranging from 35.2 to 88.2 RU, were, however, relatively low in comparison with that of a hepatocellular carcinoma cell line (Hep 3B; e-value 118 RU).
Figure 3.

hTERT protein expression after treatment with oestradiol (E2) in three human normal hepatic cell lines ((A) Hc-cells; (B) WRL-68; (C) h-Nheps) demonstrated by western blotting. The final concentration of oestradiol in the supernatant was 1, 10, and 100 nM. A concentration dependent increase in hTERT protein expression was observed in all three cell lines. hTERT protein expression of h-Nheps (C) was weaker than those in the other cell lines (A, B).
Table 2.
Elongation values (e values) of human normal hepatic cell lines measured by the telomeric repeat elongation assay
| Cell lines | e value (RU) | |
| Oestradiol (−) | Oestradiol (+)† | |
| Hc-cells | 9.5 (4.3) | 47.0 (8.2)* |
| WRL-68 | 31.3 (6.0) | 35.2 (5.5) |
| h-Nheps | 8.1 (3.5) | 88.2 (7.1)* |
Values are means (SD) of five independent assays.
†Final concentration of oestradiol in the supernatant was 10 nM.
*p<0.05 by analysis of variance.
Figure 4.
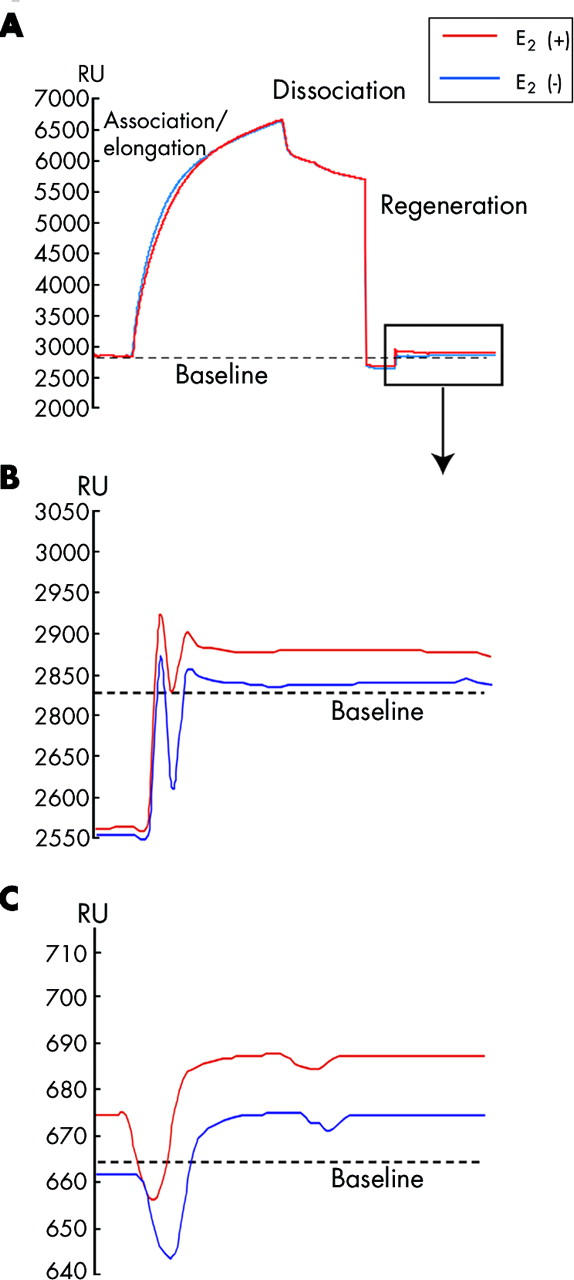
Sensorgrams of the telomeric repeat elongation assay for measuring telomerase activity in cultured hepatic cells (A, B) and tissues obtained from rats with carbon tetrachloride (CCl4) induced liver fibrosis (C). (A) Telomerase extracts (0.1 mg/ml) were injected through the flow cell (flow rate 5 μl/min) and bound to the telomeric repeat oligomers (TS-5′B). Elongation of the immobilised oligomers occurred immediately (association/elongation phase) on the sensorgram. After injection (five minute injection) of the telomerase extracts, dissociation of binding proteins was observed. Regeneration (1% sodium dodecyl sulphate 100 μl/min, one minute) was performed to remove all bound proteins. Real time monitoring was carried out throughout the association/elongation, dissociation, and regeneration phases. E2, oestradiol. (B) Difference from baseline (broken line) was defined as the e value, which was equivalent to the weight of the elongated nucleotides. An increase in the e value was observed between the points before (blue line, 14 RU) and after (red line, 66 RU) induction of oestradiol (10 nM) in Hc-cells. (C) An increase in the e value was observed between oestradiol administration (red line, 22 RU) and non-oestradiol administration (blue line, 10 RU) in male rats with CCl4 induced liver fibrosis at 24 weeks.
TRF length and senescence associated β-galactosidase staining assays in human normal hepatic cell lines on long term exposure to oestradiol
Two cell lines (Hc-cells and h-Nheps) exhibiting activation of TA by oestradiol induction were long term cultured with or without oestradiol exposure, and TRF length and senescence associated β-galactosidase staining assays were performed. WRL-68 appeared to be immortalised, judging from its telomerase activity, and therefore it was eliminated from the examination of long term oestradiol exposure. Telomere shortening was observed in both cell lines (table 3 ▶, fig 5 ▶). TRF lengths decreased with accumulated passages whereas those of oestradiol treated cells were greater than those without oestradiol exposure (fig 5 ▶). The incidence of β-galactosidase positive cells increased with accumulated passages, and was significantly decreased in oestradiol treated cells in comparison with non-treated cells at the same number of passages (figs 6 ▶, 7 ▶).
Table 3.
Mean terminal restriction fragment (TRF) length, evaluated by Southern blot analysis
| Cell lines | Mean TRF length (kb) | ||
| Oestradiol (−) | Oestradiol (+)† | ||
| Hc-cells | 10th | 4.7 (1.8) | 6.5 (1.2) |
| 20th | 3.2 (0.8) | 3.9 (0.6) | |
| 30th | 2.6 (0.2) | 3.1 (0.4) | |
| h-Nheps | 10th | 5.0 (1.4) | 6.9 (1.1) |
| 20th | 3.3 (0.6) | 4.2 (0.8) | |
| 30th | 3.0 (0.4) | 3.1 (0.5) | |
Values are mean (SD) for 5 rats.
†Final concentration of oestradiol in the supernatant was 10 nM.
Figure 5.
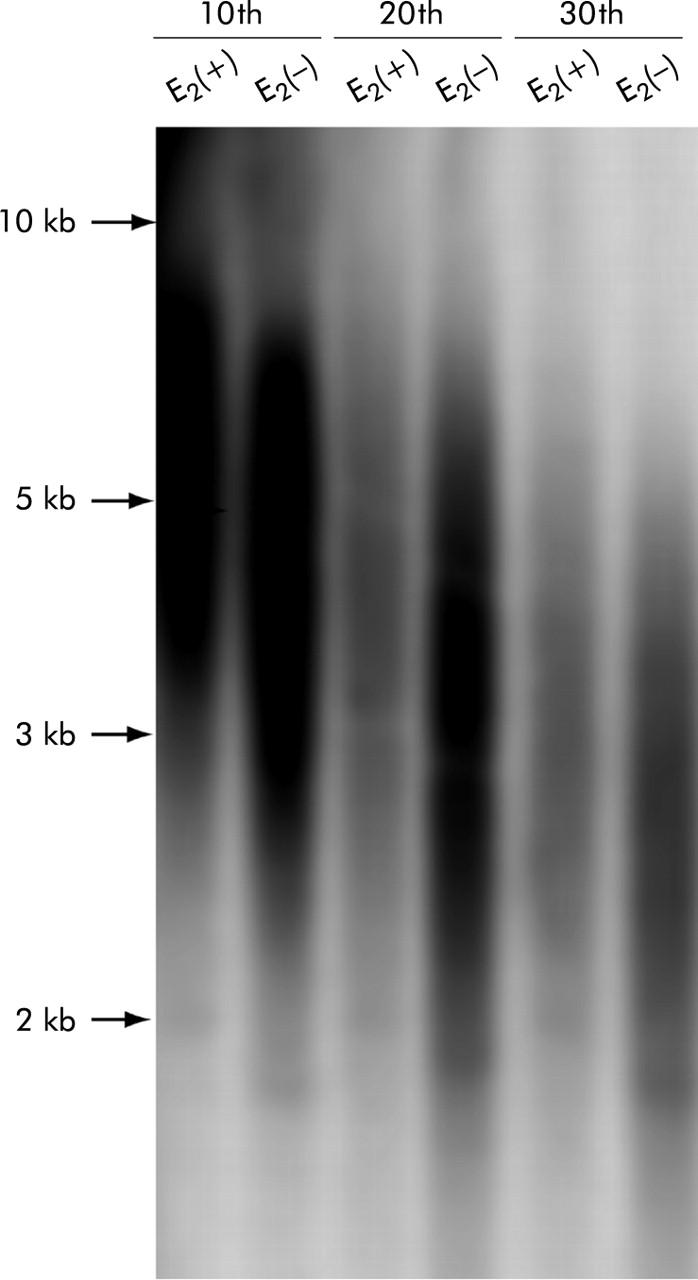
Terminal restriction fragments (TRF) in Hc-cells with or without long term oestradiol (E2) exposure by Southern blot analysis. An oligonucleotide probe (TTAGGG)4 was used. TRF lengths decreased with accumulated passages whereas oestradiol treated cells had longer telomeres than those without oestradiol exposure after the same number of passages. The sizes (kb) and positions of the markers are indicated on the left.
Figure 6.

Senescence associated β-galactosidase staining in h-Nheps with (B) and without (A) long term oestradiol exposure. Cells staining positively for senescence associated β-galactosidase showed dark granules in the cytoplasm. Positivity was frequently observed in non-oestradiol exposed cells.
Figure 7.
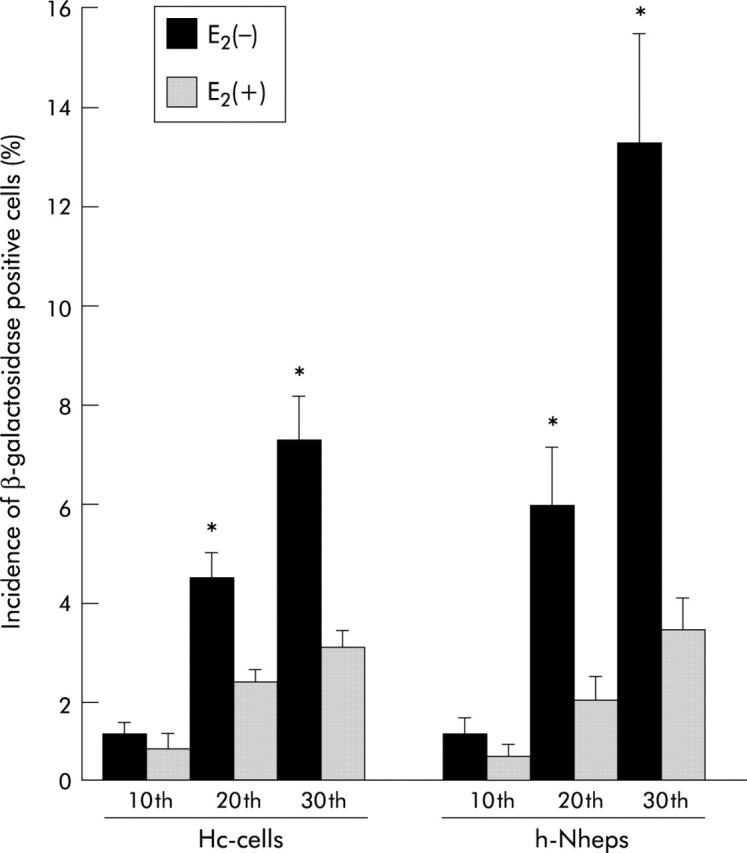
Incidence of positivity for β-galactosidase in human normal hepatic cells (Hc-cells and h-Nheps). The number of β-galactosidase positive cells increased with accumulated passages, and was significantly decreased in oestradiol (E2) treated cells in comparison with non-treated cells after the same number of passages (*p<0.05). Two researchers microscopically evaluated 500 cells five times, and determined the percentage of those staining positive for β-galactosidase.
Effects of oestradiol in the CCl4 induced cirrhosis model in the rat liver
Control livers showed only slight collagen deposition limited to the area around the central veins. Both the CCl4 alone (group A) and CCl4+oestradiol (group B) groups exhibited extensive fibrosis (P-P and P-C bridging fibrosis), infiltration of inflammatory cells, and necrosis of hepatocytes at 24 and 48 weeks. Fibrosis and architectural alterations were greater in group A livers than in group B livers (fig 8 ▶). Quantitative assessments of fibrotic areas showed significant differences between group A and group B at 24 and 48 weeks (p<0.05) (fig 9A ▶). The degree of hepatic fibrosis was more accelerated at 48 weeks than at 24 weeks. There was no significant difference in the Ki67 labelling index between the two groups (fig 9B ▶).
Figure 8.
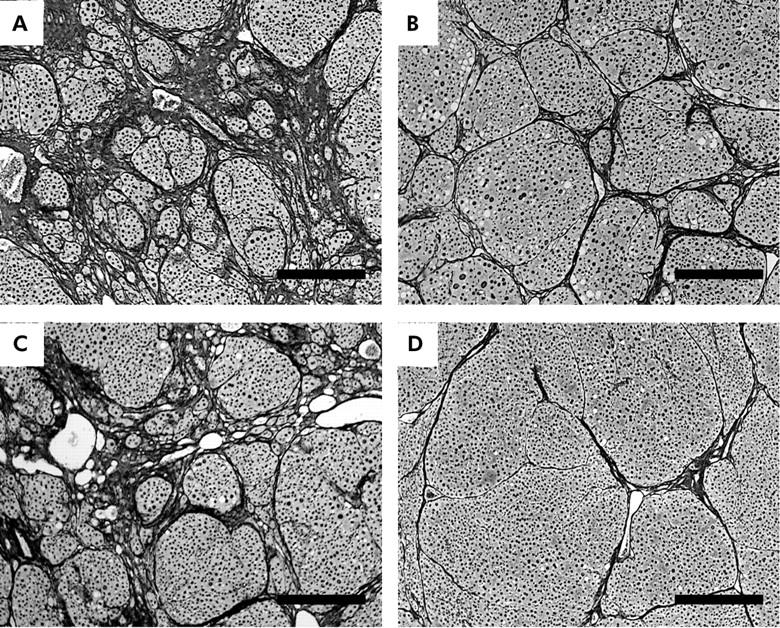
Histological appearances of carbon tetrachloride (CCl4) induced rat liver fibrosis with (B, D) and without (A, C) oestradiol administration at 24 weeks (silver impregnation, scale bar = 300 μm). The liver of a male rat administered oestradiol (B) showed less extensive fibrosis and architectural alterations than the liver of a rat without oestradiol administration (A). These alterations were more evident in females (C, D: livers of female rats without and with oestradiol administration, respectively).
Figure 9.
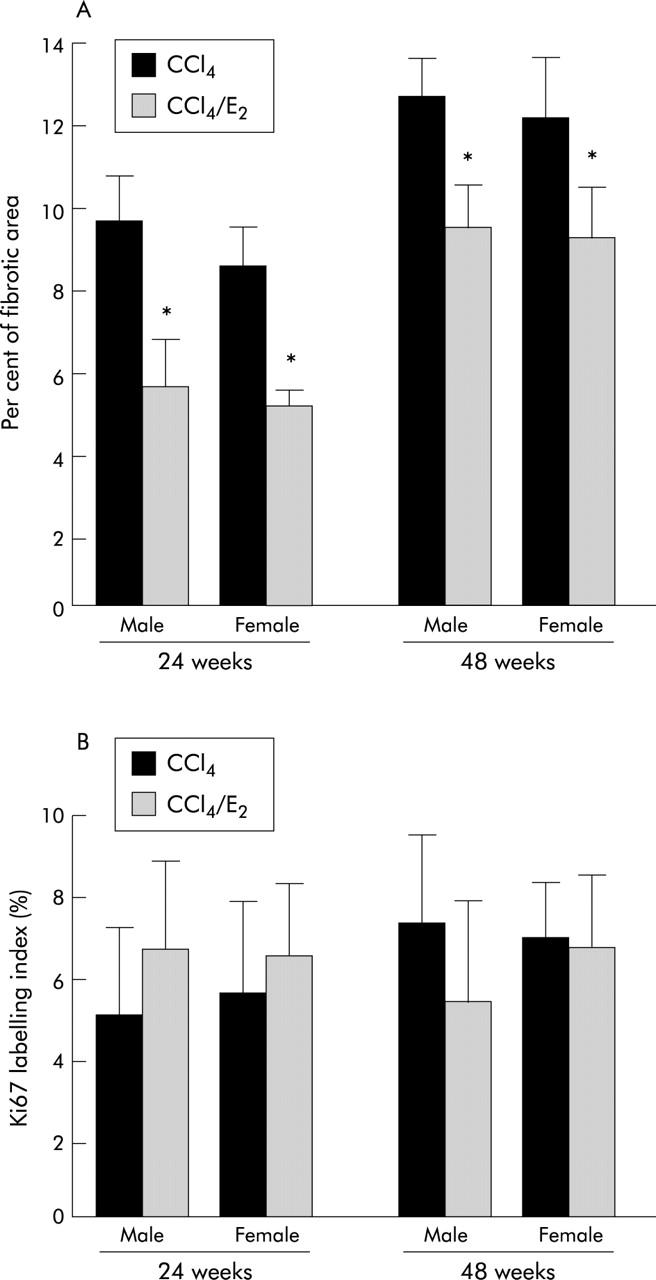
Quantitative assessments of liver fibrosis (A) and Ki67 labelling (B) in rat liver with carbon tetrachloride (CCl4) induced cirrhosis with or without oestradiol (E2) administration. Fibrotic areas were significantly decreased in oestradiol treated cells in comparison with those without oestradiol administration (p<0.05). No significant difference in the Ki67 labelling index was observed between groups with and without oestradiol administration.
In order to evaluate TA and TRF lengths, telomerase extracts and DNA were obtained from liver tissues at 24 and 48 weeks. TAs of non-treated controls and group A were extremely low, and there were no significant differences between male and female rats or between control and group A rats (table 4 ▶). TAs were significantly increased in both male and female rats of group B compared with those in Group A or in non-treated control rats (table 4 ▶, fig 4C ▶). E values in group B were slightly greater in female than in male rats but not to a significant extent (table 4 ▶, fig 4C ▶).
Table 4.
Elongation values (e values) of rat liver tissues measured by telomeric repeat elongation assay
| Rat liver | e value (RU) | |||
| Control | Group A | Group B | ||
| Male | 24 weeks | 2.8 (2.2) | 3.2 (1.5) | 20.1 (5.8)* |
| 48 weeks | 3.0 (2.0) | 3.8 (2.4) | 19.9 (9.1)* | |
| Female | 24 weeks | 1.6 (1.2) | 6.1 (3.3) | 22.7 (6.3)* |
| 48 weeks | 3.5 (2.9) | 7.4 (3.5) | 22.9 (7.4)* | |
Values are mean (SD) for five rats. We performed the TRE assay five times and calculated the mean e value for each rat.
*p<0.05 compared with the control or group A.
TRF length analysis demonstrated loss of telomeric DNA in CCl4 induced liver fibrosis. Although mean TRFs of normal rat liver were long (>20 kb), as described previously,38 extensive shortening of telomere length and a decrease in the total amount of telomeric DNA were observed as the period of CCl4 treatment was prolonged. In addition, fragments shorter than 1 kb were markedly increased in CCl4 treated rats. The source of the shorter telomeric DNA fragments (1 kb) may have been hepatocytes rendered apoptotic or necrotic by CCl4 injury. We could not evaluate mean TRF length in CCl4 induced liver fibrosis because of an abundance of shorter telomeric DNA fragments. However, the longest length of telomeric DNA was greater in female rats in group B than in those in group A at both 24 and 48 weeks (fig 10 ▶). In male rats, a difference in TRF lengths between group A and group B was confirmed at 24 weeks but was not obvious at 48 weeks. TRF lengths in female rats were greater than those in male rats.
Figure 10.
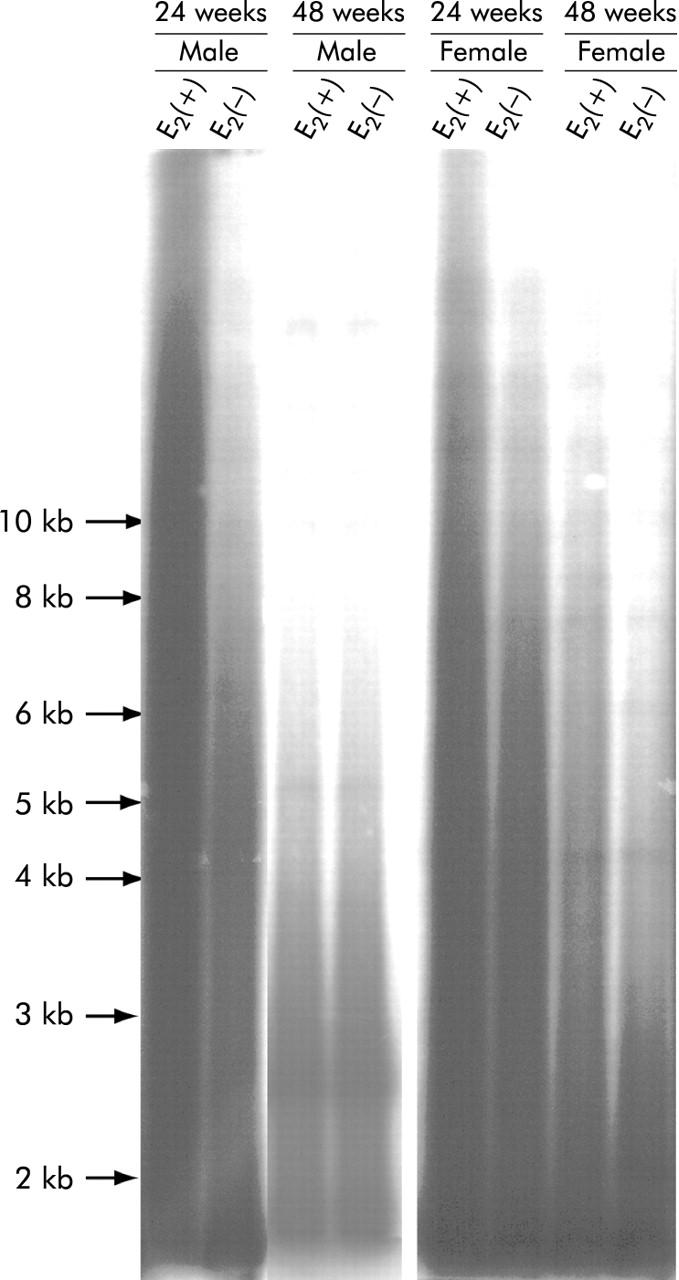
Terminal restriction fragments in rats with carbon tetrachloride (CCl4) induced liver fibrosis with and without oestradiol (E2) administration revealed by Southern blot analysis. Extensive telomere shortening and loss of total telomeric DNA were observed as the period of CCl4 treatment was prolonged. In addition, fragments shorter than 1 kb were markedly increased in CCl4 treated rats. The source of the shorter telomeric DNA fragments (1 kb) may be hepatocytes rendered apoptotic or necrotic by CCl4 injury. The longest position of the telomeric DNA was greater in male and female rats administered oestradiol than in those without oestradiol at 24 weeks. Results in female rats with and without oestradiol administration were confirmed to be identical at 48 weeks but this was not evident in male rats at the same time point.
DISCUSSION
The present study suggested that administration of exogenous oestradiol could rescue hepatocytes from extensive telomere shortening by upregulation of TA, resulting in prolongation of their life span, in vitro and in vivo.
Several studies have shown that most somatic cells, including hepatocytes, do not express TA,34,35 except for restricted cell types.36,37 Only endometrial tissue exhibits a menstrual phase dependent regulation of TA; upregulation of TA is observed during the proliferative phase (high oestradiol phase) but inhibited during the midsecretory phase (low oestradiol phase).27,29,30 Our experimental data obtained from both normal rat liver and CCl4 induced liver fibrosis failed to exhibit a significant sex related difference in TA although female rats had longer telomeres than male rats. A recent report by Cherif and colleagues38 has demonstrated age and sex related changes in telomere length in rats. The percentage of short telomeres increased with age in the liver and other organs of both male and female rats, and moreover males had shorter telomeres than females at the age of puberty. Identical sex related differences in telomere length have been reported in mouse liver, kidney, and spleen,39 and human mononuclear cells.40 These data strongly support the hypothesis that endogenous oestradiol may control hepatocyte TA and telomere length, and contribute to prolonging the lifespan of these cells.
Epidemiological data have long suggested that chronic liver disease progresses at unequal rates in the two sexes for viral hepatitis.41 Among patients with cirrhosis and hepatocellular carcinoma, the male:female ratio is within the range 2.3:1 to 2.6:1.42,43 Moreover, recent studies have suggested that elevated serum testosterone levels and an imbalanced testosterone/oestradiol rate are associated with an increased risk of hepatocellular carcinoma.44,45 Although the association was clearcut, the mechanism by which elevated levels of testosterone determine an increased incidence of hepatocellular carcinoma has been elusive.
It has been reported that both hormonal systems depending on testosterone and oestradiol are able to control TA, in vitro and in vivo.26–31 Testosterone is a negative regulator of TA. Meeker and colleagues31 demonstrated that TA is activated in the prostate and seminal vesicles of castrated male rats.31hTERT is upregulated by direct interaction with activated OR.26 Although the molecular mechanisms of the action of oestradiol have been well studied in HSC, those on hepatocytes have been unclear even though they have high affinity, low capacity, saturable, and specific OR.46,47 Our experimental data concerning the relationship between TA/telomere length and exogenous oestradiol administration have provided new insight into the progression of liver fibrosis and development of hepatocellular carcinoma. Continuous hepatocyte death and regeneration resulting in critical telomere shortening produce severe chromosomal instability, leading to massive cell death—so-called “crisis”. Severe chromosomal instability may play an important role in the development of hepatocellular carcinoma. Endogenous oestradiol may affect TA and the telomere length of hepatocytes, and may explain the relatively low incidence of hepatocellular carcinoma and end stage liver failure in female patients with chronic liver disease. Exogenous oestradiol administration appears to be a new treatment strategy that can be used as telomerase therapy for slowing the progression of chronic liver disease.
The telomerase gene therapy proposed by Depinho and colleagues2,18 is a fascinating option for restoring telomere length in patients with end stage liver failure. However, gene therapy using human telomerase RNA (human telomerase RNA component: hTERC) has a few limitations that need to be overcome before actual clinical application. As the hTERC gene is not deficient in human hepatocytes, it remains obscure whether hTERC gene delivery by adenovirus vectors would be able to increase telomerase activity and prevent critical telomere shortening in human hepatocytes as well as in hTERC deficient mice. In addition, considering the safety of vectors and the risk of cancer development by uncontrolled upregulation of TA, gene therapy would only be justified in patients with end stage liver failure (senescence or pre-crisis).48–50 However, the ideal telomerase therapy would be one that achieves early telomerase reactivation long before cirrhosis, thus preventing telomere dysfunction and genetic instability and reducing the rate of hepatocellular carcinoma development. For early start and long term telomerase therapy in patients with chronic liver disease, exogenous oestradiol administration is a more suitable therapeutic option than gene therapy. In the present study, we did not evaluate telomere related factors in HSCs that play an important role in hepatic fibrosis. Recent publications by Schnabl and colleagues51,52 suggested that exogenous hTERT expression resulting from retrovirus infection has a positive effect on hepatic fibrosis of HSCs. This was contradictory to our result suggesting that exogenous oestradiol administration would be helpful for preventing hepatic fibrosis. However, their experimental results using immortal HSCs infected with retrovirus were not simply compared with those of oestradiol administration. In fact, the effects of oestradiol administration in vitro and in vivo had already been reported.15,17 The latter studies demonstrated that the HSC phenotype was changed from activated status to quiescent status by oestradiol administration. Oestradiol exerts many actions through direct or indirect signalling pathways, and we believe that some of these may contribute to downregulation of molecules related to hepatic fibrosis.
Long term exposure to oestradiol is one of the risk factors for the development of hepatocellular carcinoma. We did not evaluate serum oestradiol levels in rats with CCl4 induced liver fibrosis. The dose of oestradiol we used was the same as that in a previous study in which exogenous oestradiol administration suppressed fibrosis in rats with dimethylnitrosamine induced liver cirrhosis.15 In that study, serum oestradiol levels were at least 200-fold higher than those in normal non-gestational rats.15 Such a high level of oestradiol may be crucial from the viewpoint of hepatocyte proliferation51 and free radical mediated DNA/RNA damage,53,54 as described previously. However, a hyperoestrogenic state (more evident in males, known as feminisation) is frequently observed in patients with chronic liver disease without hepatocellular carcinoma. In addition, an increased testosterone/oestradiol ratio is a risk factor for hepatocellular carcinoma development whereas an increased serum oestradiol level is not in itself an independent risk factor.44,45 The contribution of a high oestradiol level to hepatocellular carcinoma development thus remains debatable.
Further studies are required to determine whether a lower dose of exogenous oestradiol activates TA of hepatocytes in patients with chronic liver disease and helps rescue them from critical telomere shortening. We now intend to examine serum oestradiol levels, OR expression, TA, and telomere length in humans with chronic liver disease.
Acknowledgments
The authors wish to thank Associate Professor Kei Takahashi, Division of Mathematics, Iwate Medical University School of Liberal Arts and Sciences, for statistical analyses.
The work was supported in part by a grant from Keiryokai Research Foundation 2001-2002, and grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (No 14570156 and 15591432).
Abbreviations
HSC, hepatic stellate cells
TGF-β1, transforming growth factor-β1
M1, mortality stage 1
M2, mortality stage 2
OR, oestrogen receptor
TA, telomerase activity
TRF, terminal restriction fragment
RQ-PCR, real time quantitative-polymerase chain reaction
TRE assay, telomeric repeat elongation assay
e value, elongation value
CCl4, carbon tetrachloride
SDS, sodium dodecyl sulphate
RU, resonance unit
PBS, phosphate buffered saline
REFERENCES
- 1.Wiemann SU, Satyanarayana A, Tsahuridu M, et al. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. FASEB J 2002;16:935–42. [DOI] [PubMed] [Google Scholar]
- 2.Rudolph KL, Depinho RA. Telomeres and telomerase in experimetal liver cirrhosis. In: Arias IM, Boyer JL, Chisari FV, et al, eds. The liver: biology and pathobiology. Philadelphia: Lippincott Williams and Wilkins, 2001:1000–10.
- 3.Kitada T , Seki S, Kawakita N, et al. Telomere shortening in chronic liver diseases. Biochem Biophys Res Commun 1995;211:33–9. [DOI] [PubMed] [Google Scholar]
- 4.Miura N , Horikawa I, Nishimoto A, et al. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet Cytogenet 1997;93:56–62. [DOI] [PubMed] [Google Scholar]
- 5.Urabe Y , Nouso K, Higashi T, et al. Telomere length in human liver diseases. Liver 1996;16:293–7. [DOI] [PubMed] [Google Scholar]
- 6.Aikata H , Takaishi H, Kawakami Y, et al. Telomere reduction in human liver tissues with age and chronic inflammation. Exp Cell Res 2000;256:578–82. [DOI] [PubMed] [Google Scholar]
- 7.Arthur MJ. Fibrosis and altered matrix degradation. Digestion 1998;59:376–80. [DOI] [PubMed] [Google Scholar]
- 8.Friedman SL. Cellular networks in hepatic fibrosis. Digestion 1998;59:368–71. [DOI] [PubMed] [Google Scholar]
- 9.Schuppan D , Cho JJ, Jia JD, et al. Interplay of matrix and myofibroblasts during hepatic fibrogenesis. Curr Topic Pathol 1999;93:205–18. [DOI] [PubMed] [Google Scholar]
- 10.Martinez-Hernandez A , Amenta PS. The hepatic extracellular matrix. II. Ontogenesis, regeneration and cirrhosis. Virchows Arch A Pathol Anat Histopathol 1993;423:77–84. [DOI] [PubMed] [Google Scholar]
- 11.Fausto N , Mead JE, Gruppuso PA, et al. Effects of TGF-betas in the liver: cell proliferation and fibrogenesis. Ciba Found Symp 1991;157:165–74. [DOI] [PubMed] [Google Scholar]
- 12.Milani S , Herbst H, Schuppan D, et al. Transforming growth factors beta 1 and beta 2 are differentially expressed in fibrotic liver disease. Am J Pathol 1991;139:1221–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Roulot D , Sevcsik AM, Coste T, et al. Role of transforming growth factor beta type II receptor in hepatic fibrosis: studies of human chronic hepatitis C and experimental fibrosis in rats. Hepatology 1999;29:1730–8. [DOI] [PubMed] [Google Scholar]
- 14.Bedossa P , Peltier E, Terris B, et al. Transforming growth factor-beta 1 (TGF-beta 1) and TGF-beta 1 receptors in normal, cirrhotic, and neoplastic human livers. Hepatology 1995;21:760–6. [PubMed] [Google Scholar]
- 15.Yasuda M , Shimizu I, Shiba M, et al. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999;29:719–27. [DOI] [PubMed] [Google Scholar]
- 16.Lee KS, Buck M, Houglum K, et al. Activation of hepatic stellate cells by TGF alpha and collagen type I is mediated by oxidative stress through c-myb expression. J Clin Invest 1995;96:2461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimizu I , Mizobuchi Y, Yasuda M, et al. Inhibitory effect of oestradiol on activation of rat hepatic stellate cells in vivo and in vitro. Gut 1999;44:127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudolph KL, Chang S, Millard M, et al. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science 2000;287:1253–8. [DOI] [PubMed] [Google Scholar]
- 19.Williams EJ, Iredale JP. Liver cirrhosis. Postgrad Med J 1998;74:193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman SL. Seminars in medicine of the Beth Israel Hospital, Boston. The cellular basis of hepatic fibrosis. Mechanisms and treatment strategies. N Engl J Med 1993;328:1828–35. [DOI] [PubMed] [Google Scholar]
- 21.Alcolado R , Arthur MJ, Iredale JP. Pathogenesis of liver fibrosis. Clin Sci (Lond) 1997;92:103–12. [DOI] [PubMed] [Google Scholar]
- 22.Xu JW, Gong J, Chang XM, et al. Estrogen reduces CCL4-induced liver fibrosis in rats. World J Gastroenterol 2002;8:883–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyne J , Ratliff RL, Moyzis RK. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc Natl Acad Sci U S A 1989;86:7049–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayflick L . The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 1965;37:614–36. [DOI] [PubMed] [Google Scholar]
- 25.Chin L , Artandi SE, Shen Q, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell 1999;97:527–38. [DOI] [PubMed] [Google Scholar]
- 26.Kyo S , Takakura M, Kanaya T, et al. Estrogen activates telomerase. Cancer Res 1999;59:5917–21. [PubMed] [Google Scholar]
- 27.Kyo S , Takakura M, Kohama T, et al. Telomerase activity in human endometrium. Cancer Res 1997;57:610–14. [PubMed] [Google Scholar]
- 28.Bednarek AK, Chu Y, Aldaz CM. Constitutive telomerase activity in cells with tissue-renewing potential from estrogen-regulated rat tissues. Oncogene 1998;16:381–5. [DOI] [PubMed] [Google Scholar]
- 29.Williams CD, Boggess JF, LaMarque LR, et al. A prospective, randomized study of endometrial telomerase during the menstrual cycle. J Clin Endocrinol Metab 2001;86:3912–17. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka M , Kyo S, Takakura M, et al. Expression of telomerase activity in human endometrium is localized to epithelial glandular cells and regulated in a menstrual phase-dependent manner correlated with cell proliferation. Am J Pathol 1998;153:1985–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meeker AK, Sommerfeld HJ, Coffey DS. Telomerase is activated in the prostate and seminal vesicles of the castrated rat. Endocrinology 1996;137:5743–6. [DOI] [PubMed] [Google Scholar]
- 32.Maesawa C , Inaba T, Sato H, et al. A rapid biosensor chip assay for measuring of telomerase activity using surface plasmon resonance. Nucleic Acids Res 2003;31:E4–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dimri GP, Lee X, Basile G, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995;92:9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Lange T , DePinho RA. Unlimited mileage from telomerase? Science 1999;283:947–9. [DOI] [PubMed] [Google Scholar]
- 35.Kim NW, Piatyszek MA, Prowse KR, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266:2011–15. [DOI] [PubMed] [Google Scholar]
- 36.Taylor RS, Ramirez RD, Ogoshi M, et al. Detection of telomerase activity in malignant and nonmalignant skin conditions. J Invest Dermatol 1996;106:759–65. [DOI] [PubMed] [Google Scholar]
- 37.Hiyama K , Hirai Y, Kyoizumi S, et al. Activation of telomerase in human lymphocytes and hematopoietic progenitor cells. J Immunol 1995;155:3711–15. [PubMed] [Google Scholar]
- 38.Cherif H , Tarry JL, Ozanne SE, et al. Ageing and telomeres: a study into organ- and gender-specific telomere shortening. Nucleic Acids Res 2003;31:1576–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coviello-McLaughlin GM, Prowse KR. Telomere length regulation during postnatal development and ageing in Mus spretus. Nucleic Acids Res 1997;25:3051–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melk A , Ramassar V, Helms LM, et al. Telomere shortening in kidneys with age. J Am Soc Nephrol 2000;11:444–53. [DOI] [PubMed] [Google Scholar]
- 41.Bissell DM. Sex and hepatic fibrosis. Hepatology 1999;29:988–9. [DOI] [PubMed] [Google Scholar]
- 42.Zaman SN, Melia WM, Johnson RD, et al. Risk factors in development of hepatocellular carcinoma in cirrhosis: prospective study of 613 patients. Lancet 1985;1:1357–60. [DOI] [PubMed] [Google Scholar]
- 43.Villa E , Baldini GM, Pasquinelli C, et al. Risk factors for hepatocellular carcinoma in Italy. Male sex, hepatitis B virus, non-A non-B infection, and alcohol. Cancer 1988;62:611–15. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka K , Sakai H, Hashizume M, et al. Serum testosterone:estradiol ratio and the development of hepatocellular carcinoma among male cirrhotic patients. Cancer Res 2000;60:5106–10. [PubMed] [Google Scholar]
- 45.Yu MW, Chen CJ. Elevated serum testosterone levels and risk of hepatocellular carcinoma. Cancer Res 1993;53:790–4. [PubMed] [Google Scholar]
- 46.Porter LE, Elm MS, Van Thiel DH, et al. Characterization and quantitation of human hepatic estrogen receptor. Gastroenterology 1983;84:704–12. [PubMed] [Google Scholar]
- 47.Rossini GP, Baldini GM, Villa E, et al. Characterization of estrogen receptor from human liver. Gastroenterology 1989;96:1102–9. [DOI] [PubMed] [Google Scholar]
- 48.Dai WJ, Jiang HC. Advances in gene therapy of liver cirrhosis: a review. World J Gastroenterol 2001;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Berger A . Cirrhosis may be amenable to telomerase treatment. BMJ 2000;320:536A. [PMC free article] [PubMed] [Google Scholar]
- 50.Hagmann M . Biomedicine. New genetic tricks to rejuvenate ailing livers. Science 2000;287:1185–7. [DOI] [PubMed] [Google Scholar]
- 51.Schnabl B , Choi YH, Olsen JC, et al. Immortal activated human hepatic stellate cells generated by ectopic telomerase expression. Lab Invest 2002;82:323–33. [DOI] [PubMed] [Google Scholar]
- 52.Schnabl B , Purbeck CA, Choi YH, et al. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 2003;37:653–64. [DOI] [PubMed] [Google Scholar]
- 53.De Maria N , Manno M, Villa E. Sex hormones and liver cancer. Mol Cell Endocrinol 2002;193:59–63. [DOI] [PubMed] [Google Scholar]
- 54.Yager JD, Yager R. Oral contraceptive steroids as promoters of hepatocarcinogenesis in female Sprague-Dawley rats. Cancer Res 1980;40:3680–5. [PubMed] [Google Scholar]