

Abstract
Background/Aims: Thiazolidinediones (TZD) are a new class of oral antidiabetic drugs that have been shown to inhibit growth of some epithelial cancer cells. Although TZD were found to be ligands for peroxisome proliferators activated receptor γ (PPARγ) the mechanism by which TZD exert their anticancer effect is currently unclear. Furthermore, the effect of TZD on local motility and metastatic potential of cancer cells is unknown. The authors analysed the effects of two TZD, rosiglitazone and pioglitazone, on invasiveness of human pancreatic carcinoma cell lines in order to evaluate the potential therapeutic use of these drugs in pancreatic adenocarcinoma.
Methods: Expression of PPARγ in human pancreatic adenocarcinomas and pancreatic carcinoma cell lines was measured by reverse transcription polymerase chain reaction and confirmed by western blot analysis. PPARγ activity was evaluated by transient reporter gene assay. Invasion assay was performed in modified Boyden chambers. Gelatinolytic and fibrinolytic activity were evaluated by gel zymography.
Results: TZD inhibited pancreatic cancer cells’ invasiveness, affecting gelatinolytic and fibrinolytic activity with a mechanism independent of PPARγ activation and involving MMP-2 and PAI-1 expression.
Conclusion: TZD treatment in pancreatic cancer cells has potent inhibitory effects on growth and invasiveness suggesting that these drugs may have application for prevention and treatment of pancreatic cancer in humans.
Keywords: thiazolidinediones, pancreatic cancer, PPARγ, cancer invasiveness
Pancreatic cancer behaves aggressively, and is an important cause of cancer related mortality.1 Malignant pancreatic cancer cells are characterised by uncontrolled growth, inability to express the differentiated features of normal duct cells, and rapid invasion of adjacent tissues.2 Because of the insidious nature of its symptoms, pancreatic adenocarcinoma is often diagnosed when locally advanced and metastases are already established.3 As a consequence, available therapeutic strategies based on surgery, radiotherapy, and conventional chemotherapy are still largely unsatisfactory, showing only a minimal impact on the survival rate of patients affected by these tumours.4
Thiazolidinediones (TZD) such as pioglitazone (PGZ) and rosiglitazone (RGZ) are a new class of antidiabetic drugs which attenuate insulin resistance associated with obesity, hypertension, and impaired glucose tolerance in humans, as well as in several animal models of non-insulin dependent diabetes mellitus.5 TZD were found to be ligands for peroxisome proliferators activated receptor γ (PPARγ), a member of the nuclear receptor superfamily of ligand dependent transcription factors that is predominantly expressed in adipose tissue (although it is also expressed in other tissues at much lower levels).6,7 Recently, TZD have been shown to inhibit the growth of some epithelial cancer cell lines.8 Treatment of cultured breast and colon cancer cells with TZD resulted in a reduction of growth rate and changes in cell morphology and gene expression representative of a more differentiated state.9,10 Furthermore, human colorectal cancer cells implanted in nude mice were shown to grow more slowly in mice treated with TZD, with a 50% reduction of tumour volume.11 In addition, loss-of-function mutation of the PPARγ gene has been found in some human colon and thyroid carcinomas.12,13 As a consequence, PPARγ has become a potential molecular target for anticancer drug development, and TZD have been proposed for differentiation mediated therapy of PPARγ expressing tumours.
Although it has recently been shown that PPARγ is expressed in human pancreatic cancer and that troglitazone (the first TZD marketed for use in humans) inhibited pancreatic cell proliferation,14,15 the effect of TZD on the invasiveness and metastatic potential of pancreatic cancer cells remains unknown. Furthermore, the mechanisms by which PPARγ agonists exert their anticancer activity have not been conclusively established.
In this study we analysed the effects of TZD on the ability of pancreatic cancer cells to invade through reconstituted basal membranes and we examined the role of the matrix metalloproteinases and the plasminogen activator system in mediating the TZD induced effect.
MATERIALS AND METHODS
Materials
Most chemicals and supplies were purchased from Sigma Chemical Company (St Louis, MO, USA). Nitrocellulose and Nytran were from Schleicher and Schuell Inc (Keene, NH, USA). Agarose, trypsin, all restriction endonucleases, DNA modifying enzymes, and tissue culture media were purchased from Gibco BRL (New Brunswick, NJ, USA). Fetal bovine serum (FBS) was from Hyclone Laboratories (Logan, UT, USA). [Methyl-3H]thymidine ([3H]TdR) and D-threo-[dichloroacetyl-1,2-14C]-chloramphenicol were purchased from New England Nuclear (Boston, MA, USA). Bisphenol A diglycidyl ether (BADGE) was from Fluka (Milwaukee, WI, USA). GW9662 was from Cayman Chemical (Ann Arbor, MI, USA). 15d-PGJ2 was from Biomol (Plymouth Meeting, PA, USA). Rosiglitazone and pioglitazone were from Smith-Kline Beecham Pharmaceuticals (Welwyn, UK, USA) and Takeda Chemicals (Tokyo, Japan), respectively.
Tissue sample and cell cultures
Tumour samples were obtained from surgical specimens. The ethics committee of our institution approved the protocol for the study, and informed written consent was obtained from all patients. Tissues were immediately frozen in liquid nitrogen after collection and stored at −80°C.
The pancreatic adenocarcinoma cell lines PANC-1, MIA PaCa-2, BxPC-3, CAPAN-2, CAPAN-1, AsPC-1, HPAC, PL-45, and SU.86.86 were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). The PK-1 cell line was a gift from Dr A Scarpa. The PANC-1, MIA PaCa-2, HPAC, PL-45, CAPAN-1, CAPAN-2, and PK-1 cells were maintained in Dulbecco’ s modified Eagles’ medium (DMEM; GIBCO Laboratories, Grand Island, NY, USA) containing 10% FBS (Gibco). BxPC-3, AsPC-1 and SU.86.86 were maintained in RPMI-1640 (Gibco) containing 10% FBS. Culture medium was supplemented with 50 U/mL penicillin and 50 μg/mL streptomycin. All experiments were performed in phenol red free media.
Growth assay
Cell were plated in 30 mm culture dishes at the density of 50 000 cells/well in the presence of medium containing 10% FBS. After an attachment period of 24 hours, cells were incubated for different periods of time in serum free medium in presence or absence (control) of TZD at the appropriate concentration. At the indicated times, cells were harvested by trypsinisation. Cells were resuspended in phosphate buffered saline to ensure a single cell suspension. Viable cells were countered in a hemocytometer by trypan blue exclusion. Triplicate or quadruplicate wells were analysed for each experimental point.
DNA synthesis assay
DNA synthesis was evaluated by the amount of [methyl-3H]thymidine ([3H]TdR) incorporated into trichloroacetic acid precipitated materials. Cells were seeded at the density of 5×104 cells/24 well plates and incubated in complete medium until they became 70% confluent. After 24 hours‘ incubation in serum free medium, cells were treated with TZD for 42 hours and then pulsed for six hours with 1.0 μCi/ml [3H]TdR (6.7 Ci/mmol). Sextuplicate determinations were used for each experimental point.
Invasion assay
The ability of cells to invade through a Matrigel coated filter was measured in Boyden chambers. Polyvinylpyrrolidone free polycarbonate filters (pore size 8 μm) were coated with basement membrane Matrigel (200 μg/filter) (Collaborative Research Inc, Bedford, MA, USA) as described in the standard protocol.16 Confluent cells were serum starved for 24 hours and then exposed to TZD or vehicle for one hour. The cells were then washed, trypsinised, suspended in serum free medium with or without TZD at the density of 1×105, and placed in the upper chambers. DMEM containing 1% FBS was placed in the lower compartments of the Boyden chambers. Chambers were incubated for six hours in 5% CO2/95% air at 37°C. The filters were fixed in methanol and were stained with haematoxylin and eosin. Cells from various areas of the lower surface were counted using a computerised video image analysis system (Leica Quantimet Q500MC, Leica Cambridge Ltd, Cambridge, UK). Each assay was performed in triplicate.
Proteins extract and western blot
Whole cell proteins were extracted from each adenocarcinoma cell line. Cells were cultured in the presence or absence of test agents and were homogenised in Laemmli buffer.17 Nuclear proteins were isolated from treated and untreated cells based on micropreparation methods.18 Nuclear and whole cell extracts (40 μg protein) were fractionated in 12% sodium dodecyl-sulfate-polyacrylamide gel electrophoresis and electroblotted onto nitrocellulose filters. Proteins were detected by incubating the filters with a mouse antihuman PPARγ monoclonal antibody (1:500) (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Detection of the protein bands was performed using the Amersham ECL kit (Arlington Heights, IL, USA).
RNA extraction and RT-PCR
Total RNA was extracted from cultured cells and tissue samples by the guanidinium-phenol-chloroform methods of Chomczynski and Sacchi19 with minor modifications.20 One microgram of RNA from tissue or tumour cells was reversed transcribed with Molony murine leukaemia virus (MMLV) reverse transcriptase (Life Technologies Inc, Paisley, UK) at 42°C for 60 minutes in a 20 μl mixture in the presence of random hexamers. The nucleotide bases used in the study were previously described.21 Two μl of a reverse transcribed mixture was subjected to PCR in a 20 μl reaction solution (10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2.0 mM MgCl2, 0.01% gelatin, 20 mM deoxynucleotide triphosphate, 0.5 units of Taq polymerase (Life Technologies Inc, Paisley, UK), and 0.25 pmol of primers). Twenty five cycles of reaction at 94°C for 50 seconds, 60°C for 45 seconds, and 72°C for 90 seconds were carried out by DNA thermal cycler (Perkin-Elmer Cetus Norwalk, CT, USA). Efficiency of RT was controlled in each samples by PCR amplification of human β2 microglobulin (5′-GCAAAAGATGAGTATGCCTG–3′, 5′- TTCACTCAATCCAAATGCGG –3′).
Northern blot and RNase protection assay
Twenty micrograms of total RNA for each sample were electrophoresed in a 1% agarose 3% formaldehyde gel and transferred onto a nylon membrane. Filters were prehybridised and then hybridised overnight at 42°C with the complementary PPARγ cDNA probe.22 The probe was labeled with [32P]deoxycytidine 5′-triphosphate to a specific activity of 2.5×108 cpm/μg DNA by using a random primer kit (Amersham, Little Chalfont, UK). Equal loading of the samples was checked by reprobing the blots with a probe encoding the ribosomal protein 36B4.23 Levels of MMP-2 and PAI-1 mRNA were determined by RNase protection assay (RPA) as previously detailed.24 “Antisense” (complementary to mRNA) RNA probes were obtained by run off transcription of pGEM1 (Promega, Madison, WI, USA) containing the 1300 bp EcoRI-BglII cDNA fragments of pK121,25 kindly provided by Dr K Tryggvason, coding for human gelatinase A (MMP-2). The PAI-1 probe was generated by oligo-(dT)-primed reverse transcription of total cell RNA prepared from human placenta, with subsequent amplification using specific oligodeoxyribonucleotide primers.26 The HindIII-HindIII 110-bp cDNA fragment encoding for human glyceraldehydes phosphatase dehydrogenase (GAPDH) was used as a “housekeeping gene”.
Transient transfection of culture cells
Cells were transfected at the density of 5×105 cells/60 mm dish by calcium phosphate precipitation with 2.5 μg of peroxisome proliferator response element (ARE)3-tk-luciferase reporter plasmid (containing three copies of the PPRE from the adipocyte lipid binding protein (aP2) gene ligated to a herpes simplex thymidine kinase promoter upstream of a luciferase gene),27 5 μg of human PPARγ expression plasmid, and 5 μg of pSV2CAT (containing SV40 early promoter and enhancer sequences that drives a chimeric chloramphenicol acetyl transferase (CAT) gene) as an internal control. Luciferase assay was performed based on the original protocol of de Wet et al.28 The number of relative light units with a three second delay and a 30 second incubation were measured by Sirius1 luminometer (Berthold Detection System, Pforzheim, Germany). CAT activity was measured as described previously.29 The conversion of chloramphenicol to its acetylated products was quantified on Ambis beta scanner (Ambis System, San Diego, CA, USA).
Zymography
The gelatinolytic and fibrinolytic activity was determined in the supernatants of TZD treated and untreated pancreatic cell lines. For this purpose, cells were cultured for 24 hours in serum free medium, washed twice, and finally treated with TZD for further 24 hours. Supernatants were collected, concentrated using centrifugal filter devices (Centricon) (Millipore Corp, Bedford, MA, USA), and protein content was determined using BCA Protein Assay Reagent (Pierce, Rockford, IL, USA). Equal amounts of protein (5 μg/lane) were mixed with SDS sample buffer without reducing agents and incubated for 20 minutes at 37°C. For gelatinolytic activity assay, samples were separated on polyacrylamide gels containing 1 mg/ml gelatin. After electrophoresis, gels were stained for one hour in a 45% methanol/10% acetic acid containing Coomassie brilliant blue G250 and destained. For fibrinolytic activity assay, aliquots of culture medium were subjected to sodium dodecyl sulfate-polyacrylamide slab gel electrophoresis (10%) and migrated proteins were transferred onto 0.45 μm pore size nitrocellulose filter as previously described.30 Zymographic analyses were performed in at least three independent experiments.
Statistical methods
Data were expressed as mean (standard deviation). Statistical correlation of data were checked for significance by ANOVA and paired Student’s t test. The corresponding probability (p) is given.
RESULTS
PPARγ expression and activity in human pancreatic adenocarcinoma and cultured cell lines
The expression of PPARγ was detected by reverse transcriptase polymerase chain reaction (RT-PCR) in a number of human pancreatic carcinoma and in well characterised pancreatic cancer cell lines. As shown in table 1 ▶ and figure 1 ▶, PPARγ mRNA was detected in 23 of 42 pancreatic adenocarcinomas and in seven of 10 cell lines of varying differentiation (fig 2A ▶). A further characterisation by northern and western analysis showed that Panc-1, BxPC3, and Capan-2 cell lines expressed both PPARγ mRNA and protein, whereas HPAC, PL-45, and SU 86-86 cell lines did not (fig 2B and 2C ▶ ▶). All cells lines expressed RXRα, the obligatory heterodimerisation partner for PPARγ (not shown).
Table 1.
PPARγ expression in human pancreatic adenocarcinoma
| Patient | Sex | Age (years) | Histological grade | TNM | |||||
| T | N | M | Stage | PPARγ | |||||
| 1 | M | 68 | Well | 1a | 0 | 0 | I | + | |
| 2 | M | 53 | MCT | 2 | 0 | 0 | I | + | |
| 3 | F | 45 | Well | 1a | 0 | 0 | I | − | |
| 4 | F | 71 | Moderately | 2 | 0 | 0 | I | − | |
| 5 | M | 77 | Moderately | 2 | 0 | 0 | I | + | |
| 6 | F | 55 | Moderately | 2 | 0 | 0 | I | − | |
| 7 | M | 43 | MCT | 1a | 0 | 0 | I | − | |
| 8 | M | 76 | Well | 3 | 0 | 0 | II | + | |
| 9 | F | 68 | Moderately | 3 | 0 | 0 | II | + | |
| 10 | F | 75 | Moderately | 3 | 0 | 0 | II | + | |
| 11 | F | 65 | Moderately | 3 | 0 | 0 | II | + | |
| 12 | M | 70 | Well | 3 | 0 | 0 | II | − | |
| 13 | M | 77 | Moderately | 3 | 0 | 0 | II | − | |
| 14 | M | 73 | Moderately | 3 | 0 | 0 | II | + | |
| 15 | F | 61 | Moderately | 3 | 0 | 0 | II | + | |
| 16 | F | 80 | Well | 1b | 1 | 0 | III | − | |
| 17 | F | 77 | Moderately | 2 | 1 | 0 | III | − | |
| 18 | M | 55 | Moderately | 2 | 1 | 0 | III | + | |
| 19 | M | 53 | Well | 3 | 1 | 0 | III | + | |
| 20 | M | 66 | Moderately | 3 | 1 | 0 | III | − | |
| 21 | F | 64 | Moderately | 2 | 1 | 0 | III | + | |
| 22 | M | 62 | Well | 3 | 1 | 0 | III | − | |
| 23 | M | 72 | Moderately | 2 | 1 | 0 | III | + | |
| 24 | F | 57 | Moderately | 2 | 1 | 0 | III | + | |
| 25 | F | 49 | Moderately | 2 | 1 | 0 | III | − | |
| 26 | M | 56 | Well | 3 | 1 | 0 | III | − | |
| 27 | M | 81 | MCT | 3 | 1 | 0 | III | + | |
| 28 | M | 59 | Moderately | 3 | 1 | 0 | III | − | |
| 29 | M | 57 | Well | 2 | 1 | 0 | III | + | |
| 30 | M | 67 | Moderately | 3 | 1 | 0 | III | + | |
| 31 | F | 69 | Moderately | 2 | 1 | 1 | LYM | IV | − |
| 32 | F | 66 | Poorly | 2 | 1 | 1 | HEP,LYM | IV | − |
| 33 | M | 49 | Moderately | 3 | 1 | 1 | HEP | IV | − |
| 34 | M | 68 | Moderately | 3 | 1 | 1 | LYM | IV | + |
| 35 | M | 65 | Poorly | 3 | 1 | 1 | HEP, LYM | IV | + |
| 36 | F | 79 | Moderately | 3 | 1 | 1 | HEP | IV | + |
| 37 | M | 71 | Moderately | 3 | 1 | 1 | LYM | IV | − |
| 38 | F | 67 | Moderately | 3 | 1 | 1 | HEP | IV | + |
| 39 | M | 73 | Poorly | 3 | 1 | 1 | HEP, LYM | IV | − |
| 40 | M | 56 | Moderately | 3 | 1 | 1 | HEP | IV | − |
| 41 | F | 71 | Moderately | 3 | 1 | 1 | HEP | IV | + |
| 42 | M | 53 | Poorly | 3 | 1 | 1 | LYM | IV | + |
Total RNA was extracted from tissue samples by guanidinium-phenol-chloroform method as described in Methods. One microgram of total RNA was reverse transcribed using random hexamers and amplified by polymerase chain reaction using specific primers for PPARγ. The reverse transcription polymerase chain reaction products were electrophoresed on an ethidium bromide containing agarose gel.
MCT, mucinous cyst adenocarcinoma; T1, tumour limited to the pancreas; T1a, ⩽2 in greatest dimension; T1b, >2 in greatest dimension; T2, tumour extended directly to any of the following: duodenum, bile duct, or peripancreatic tissue; T3, tumour extended directly to any of the following: stomach, spleen, colon, or adjacent large vessel; N0, no regional lymph node metastasis; N1, regional lymph node metastasis; M0, no distant metastasis; M1, distant metastasis; HEP, hepatic metastasis; LYM, distant lymph node metastasis.
Figure 1.

PPARγ expression in human pancreatic adenocarcinoma. One microgram of total RNA extracted from 16 representative human pancreatic adenocarcinomas was reverse transcribed using random hexamers and amplified by polymerase chain reaction using specific primers for PPARγ and for β2-microglobulin (β2-M) as described in Methods. The reverse transcription polymerase chain reaction products were electrophoresed on ethidium bromide containing agarose gel. Numbers correspond to patient number in table 1 ▶.
Figure 2.
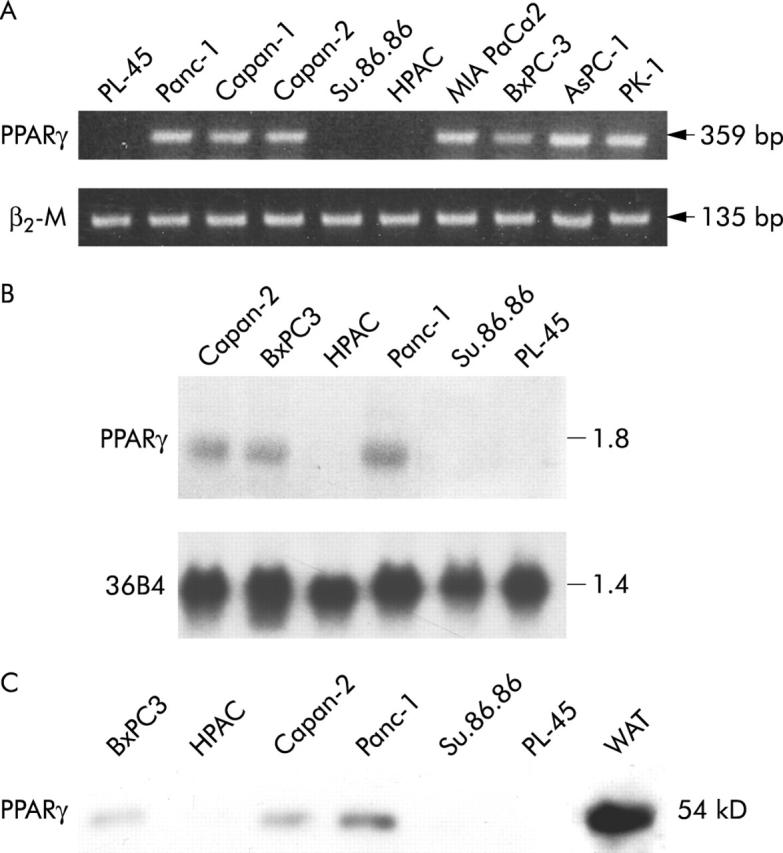
PPARγ expression in cultured human pancreatic adenocarcinoma cells. (A) RT-PCR analysis of PPARγ in 10 pancreatic cancer cell lines. One microgram of total RNA was reverse transcribed using random hexamers and amplified by polymerase chain reaction using specific primers for PPARγ and β2-microglobulin (β2-M). The reverse transcription polymerase chain reaction products were electrophoresed on ethidium bromide containing agarose gel. (B) Northern blot analysis of total RNA (20 μg) extracted from three PPARγ expressing (Panc-1, BxPC-3, Capan-2) and three non-expressing (HPAC, SU.86.86, PL-45) cell lines. Denaturated RNA sample were electophoresed in 1% agarose-3% formaldehyde gel and transferred to a nylon membrane. Filters were prehybridised and then hybridised with the complementary PPARγ and 36B4 cDNA probes as described in Methods. (C) Levels of immunoreactive PPARγ in pancreatic cancer cells. Fifty micrograms of nuclear protein extracts were fractionated by sodium dodecyl sulfate-polyaclylamide gel electrophoresis and transferred to nitrocellulose paper. Receptor proteins were detected by incubating the filter with specific anti-PPARγ antibody. Total protein extracts from human white adipose tissue (WAT) was used as positive control.
PPARγ transactivation in pancreatic cancer cell lines was monitored by activity of transfected ARE-73-tk-luc reporter (fig 3 ▶). Similarly to the natural PPARγ agonist, 15d-PGJ2, both RGZ and PGZ induced reporter activity about two to fivefold over control in PPARγ expressing cells, but neither 15d-PGJ2 nor TZD had any effect on luciferase activity in PPARγ non-expressing cells.
Figure 3.

PPARγ transcriptional activity in human pancreatic adenocarcinoma cells. After overnight attachment, cells were transfected with ARE-73-tk-luciferase reporter plasmid and pSV2-CAT as internal control for transfection efficiency. Twenty four hours after transfection cells were treated with 20 μM of TZD (RGZ or PGZ) or 5 μM of 15d-PGJ2 or vehicle (control). Twenty four hours after treatment the cells were harvested for luciferase and CAT assay as described in Methods. The data are expressed as mean (SD) for three or four replicate experiments performed in triplicate; *denotes statistical significance (p<0.01 or higher degree of significance) versus control.
Effect of TZD on growth and invasiveness of human pancreatic cancer cells
The effects of TZD on the ability of PPARγ expressing and non-expressing cells to invade through reconstituted basement membranes were analysed using Matrigel coated invasion chambers as described in Methods. Incubation with 10 μmol/l of RGZ or PGZ inhibited matrix invasiveness of both PPARγ expressing and non-expressing cancer cells (fig 4 ▶). Cell treatment with clofibric acid (1 mmol/l) and L165041 (1 μmol/l), which are potent activators of the PPARα and PPARδ isoforms respectively, but weak activators of PPARγ,27 had negligible effects on the ability of pancreatic cancer cells to invade Matrigel.
Figure 4.
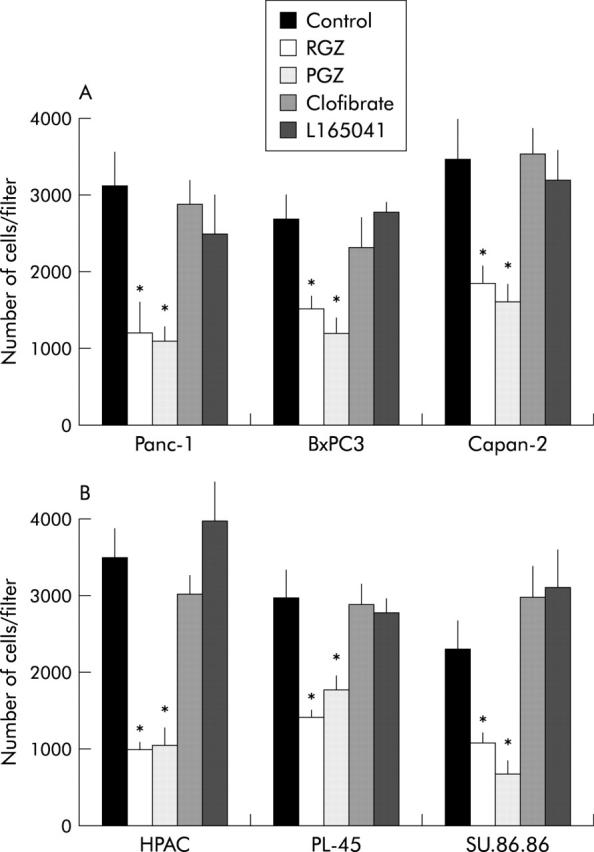
Thiazolidinediones inhibit the invasiveness of cultured pancreatic cancer cells independently of PPARγ. Confluent cells were serum starved for 24 hours and then exposed to the different receptor ligands or vehicle for one hour. Cells were then trypsinised, resuspended in serum free medium with or without ligands (20 μM TZD or 1 mM clofibric acid or 1 μM L165041), and placed in the upper compartments of Boyden chambers. After six hours’ incubation polycarbonate filters were fixed in methanol and stained with haematoxylin and eosin. Cells were then counted with a computerised video image system. (A) PPARγ expressing cell lines. (B) PPARγ non-expressing cell lines. The mean (SD) of six independent experiments performed for each in duplicate are shown; *denotes statistical significance (p<0.03 or higher degree of significance) versus vehicle treated controls.
The dose dependency of the TZD induced inhibition of cell invasiveness was further characterised in Panc-1 and in HPAC cells (fig 5 ▶). The two TZD showed a clear dose dependent inhibition of cell invasiveness and display a similar potency at all concentration used.
Figure 5.

Thiazolidinediones inhibit the invasiveness of cultured pancreatic cancer cells in a dose dependent manner. Confluent cells were serum starved for 24 hours and then exposed to TZD or vehicle (RGZ or TGZ) for one hour. Cells were then trypsinised, resuspended in serum free medium with or without TZD at the concentration indicated, and placed in the upper compartments of Boyden chambers. After six hours’ incubation polycarbonate filters were fixed in methanol and stained with haematoxylin and eosin. Cells were then counted with a computerised video image system. (A) A representative picture showing inhibition of invading cells after treatment with 20 μM of TZD. Original magnification ×200. (B) TZD treatments inhibit the invasiveness of pancreatic cancer cells in a dose dependent manner. The mean (SD) of six independent experiments performed for each in duplicate are shown; *denotes statistical significance (p<0.05 or higher degree of significance) versus vehicle treated controls.
As recent studies have shown that troglitazone, the first TZD market for use in humans, inhibits cancer cell growth independently of PPARγ expression,31 we further investigated the effects of RGZ and PGZ on pancreatic cancer cell growth. Incubation with both RGZ and PGZ resulted in a dose dependent growth inhibition of the three PPARγ expressing cell lines, but had no significant effect on PPARγ non-expressing cells (fig 6A and 6B ▶ ▶). The effect of TZD on the kinetics of DNA synthesis was also studied (fig 7 ▶). Twenty four hours of TZD incubation induced a significant reduction of [3H]TdR incorporation in PPARγ expressing cells (fig 7A ▶). In contrast RGZ and PGZ had no effect on the DNA synthesis in HPAC, PL45, and SU 86-86 cells (fig 7B ▶). In order to confirm that the effects of TZD on pancreatic cancer cell growth were dependent on PPARγ expression, we treated Panc-1 cells with two synthetic PPARγ antagonists, bisphenol A diglycidyl ether (BADGE) and GW9662.32,33 Both antagonists did not block the TDZ inhibitory effect on matrix invasiveness, but completely abrogated TDZ induced growth arrest in this PPARγ expressing cell line (fig 8 ▶).
Figure 6.
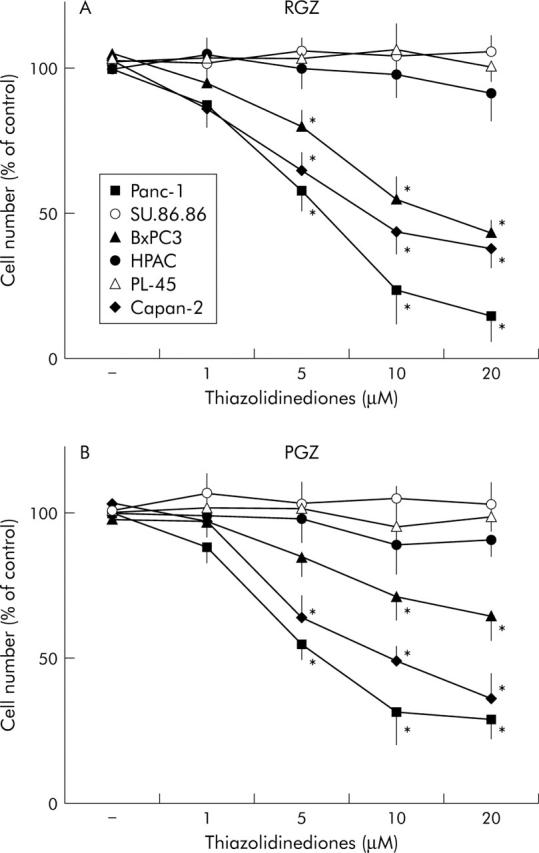
Effect of TZD treatment on anchorage dependent growth in human pancreatic adenocarcinoma cells. Cells were counted after 72 hours of incubation with TZD at appropriate concentrations. Control cells received vehicle alone. After trypsinisation cells were resuspended in phosphate buffer saline and counted in a hemocytomer as described in Methods. (A) Treatment with rosiglitazone (RGZ). (B) Treatment with pioglitazone (PGZ). PPARγ non-expressing cell lines. Cell number was expressed as percentage of untreated controls. The mean (SD) of six independent experiments each performed in duplicate or triplicate are shown; *denotes statistical significance (p<0.05 or higher degree of significance) versus control.
Figure 7.
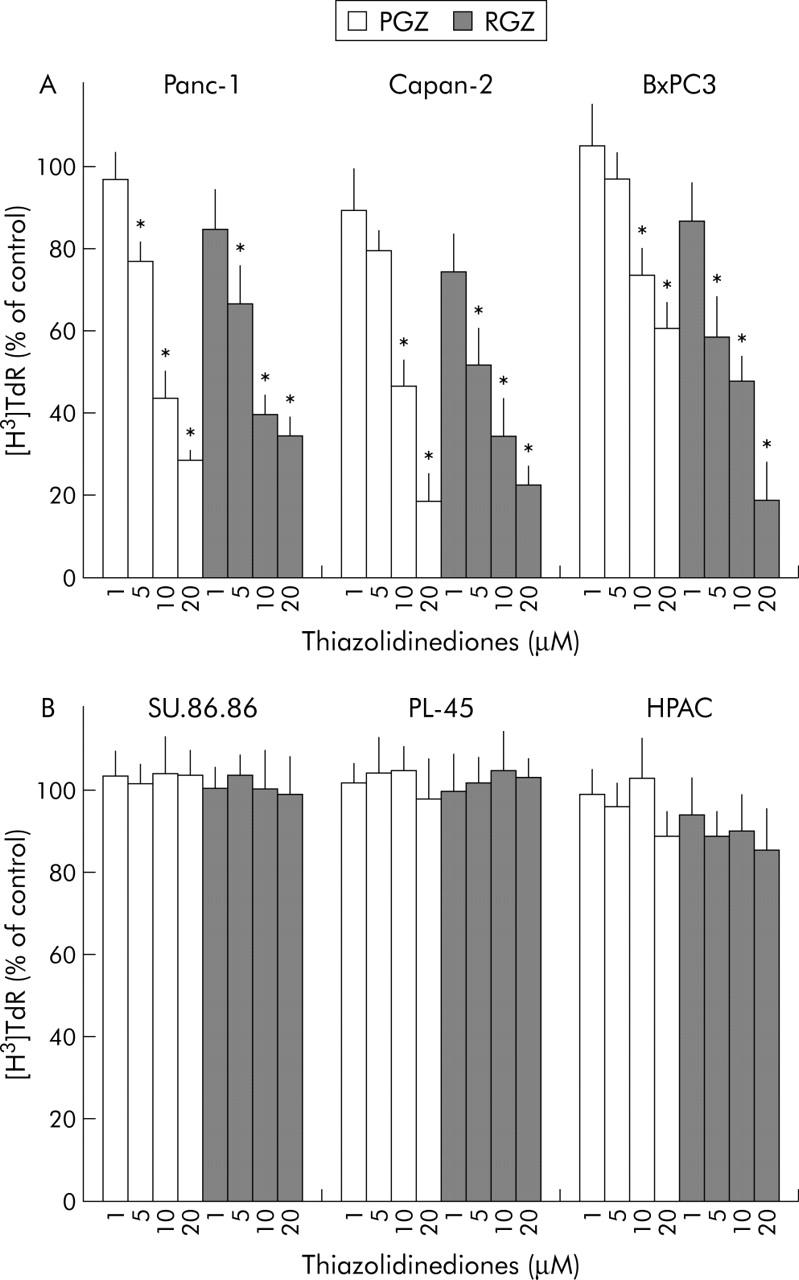
Effect of TZD treatment on DNA synthesis in human pancreatic adenocarcinoma cells. [3H]TdR incorporation: subconfluent cells were incubated in serum free/insulin free medium for 24 hours and then treated with increased concentrations of TZD (RGZ or PGZ) or their vehicle for 42 hours. After a six hour pulsing period with [3H]TdR (1.0 μCi/mL; see Methods for details) the cells were harvested. (A) PPARγ expressing cell lines. (B) PPARγ non-expressing cell lines. Data were expressed as percentage of untreated controls. The mean (SD) of six independent experiments each performed in triplicate are shown; *denotes statistical significance (p<0.05 or higher degree of significance) versus control.
Figure 8.

Effects of PPARγ antagonists on growth and invasiveness of Panc-1 cells. For the invasion assay confluent Panc-1 cells were serum starved for 24 hours and then exposed to 20 μM RGZ or vehicle with or without PPARγ antagonist (20 μM BADGE or 10 μM GW9662) for one hour. Cells were then trypsinised, resuspended in serum free medium with or without RGZ and PPARγ antagonists, and placed in the upper compartments of Boyden chambers. After six hours’ incubation polycarbonate filters were fixed in methanol and stained with haematoxylin and eosin. Cells were then counted with a computerised video image system. Cells growth was evaluated by cell counting after 72 hours of incubation with 20 μM RGZ with or without PPARγ antagonists (20 μM BADGE or 10 μM GW9662). Control cells received vehicle alone. After trypsinisation cells were resuspended in phosphate buffer saline and counted in a hemocytomer as described in Methods. Cells number was expressed as percentage of untreated controls. The mean (SD) of six independent experiments performed for each in duplicate are shown; *denotes statistical significance (p<0.01 or higher degree of significance) versus vehicle treated controls; °denotes statistical significance (p<0.02) versus RGZ treated cells.
Effect of TZD treatment on expression and activation of MMPs, and plasminogen activator system
As metalloproteases (MMPs) and the urokinase type plasminogen activator (uPA) system have been shown to play a significant role in pancreatic tumour cell invasion,34,35 we analysed gelatinolytic and fibrinolytic activity in the conditioned medium of TZD treated and untreated pancreatic cancer cells by gel zymography. As shown in figure 9 ▶, a 72 KDa band of lysis, corresponding to the latent MMP-2 was found in the conditioned medium of all pancreatic cancer cell lines whereas a faint 62 KDa band, corresponding to the activated MMP-2 was evident only in Panc-1 cells. A significant reduction of MMP-2 activity was demonstrated after 24 hour treatment with both RGZ and PGZ in conditioned medium of both PPARγ expressing and non-expressing pancreatic cancer cells. As previously demonstrated,35 PPARγ non-expressing cells, HPAC and SU.86.86, display MMP-9 related gelatinolitic activity. TZD treatment had negligible effects on latent (94 kDa) and activated (88 kDa and 77 kDa) MMP-9 in both cell lines (fig 9 ▶). Similarly the slight MMP-9 activity in BxPC3 and in Capan-2 cells was unaffected by TZD treatment. Time course analysis of MMP-2 mRNA expression was studied by RT-PCR (fig 11A ▶) and RPA (fig 11B ▶) in Panc-1 and HPAC cell lines. A significant reduction of MMP-2 transcript levels after TZD treatment occurred in these representative cell lines.
Figure 9.
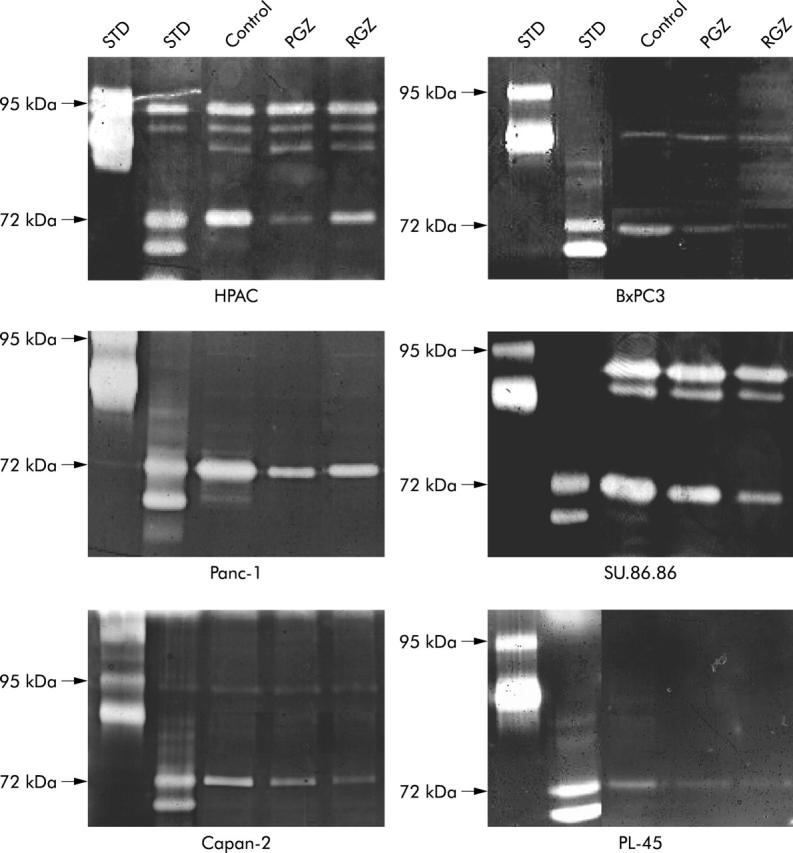
Gelatinolytic activity of conditioned medium of TZD treated pancreatic cancer cell. Cells were starved in serum free medium for 24 hours, washed, and then cultured in serum free medium with 20 μM of TZD (RGZ or PGZ) or their vehicle for further 24 hours. Supernatants were then collected and concentrated. Equal amounts of proteins (5 μg) were separated on gelatine-containing polyacrylamide gel as described in Methods. Zones of enzymatic activity are visible as bright bands. Both the 72 kDa band representing pro-MMP-2 and the 62 kDa band corresponding to active MMP-2 are reduced in supernatants of cancer cells after TZD treatment. Both latent (94 kDa) and active (88 kDa and 77 kDa) MMP-9 bands were unchanged after TZD treatment. STD, molecular standard. A representative of three independent experiments performed in duplicate, yielding similar results, is shown.
Figure 11.
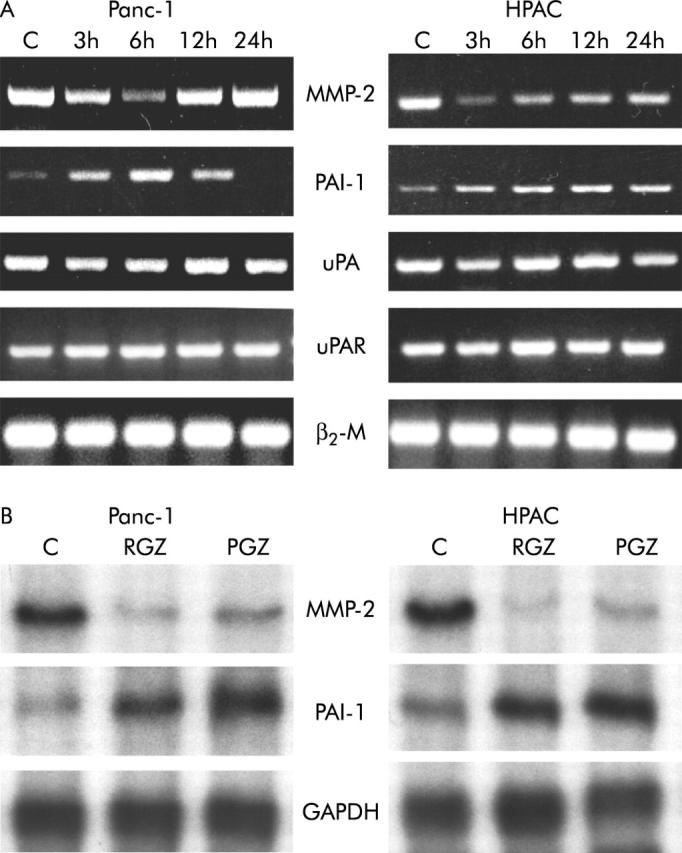
Expression of MMP-2 and uPA system genes after TZD treatment. After a 24 hour culture in serum free medium, cells were incubated with or without 20 μM of RGZ or PGZ for the indicated time intervals. (A) RT-PCR. One microgram of total RNA was reverse transcribed using random hexamers and amplified by polymerase chain reaction using specific primers. The RT-PCR products were electrophoresed on agarose gels containing ethidium bromide. (B) RNase protection assay. Ten micrograms of RNA were hybridised with antisense RNA probes specific for MMP-2, PAI-1, and GAPDH. Autoradiographic exposure was 24 hours for MMP-2 and 16 hours for PAI-1 and GAPDH. TZD inhibited MMP-2 and induced PAI-1 gene expression in both Panc-1 and HPAC cells. A representative of three independent experiment performed in triplicate, yielding similar results is shown.
In order to examine the effects of TZD on the activity of the plasminogen activator system pancreatic cancer cells were treated with RGZ or PGZ for 24 hours and plasmin dependent fibrinolytic activity was analysed in the supernatants of unstimulated and stimulated cells. As illustrated in figure 10 ▶, a single application of TZD reduced the level of fibrinolytic activity in all cell lines. In addition, expression studies by semiquantitative RT-PCR and RPA showed that TDZ increased the expression of PAI-1 in both cell lines but no effect on uPA and uPAR occurred (fig 11A and 11B ▶ ▶).
Figure 10.

Fibrinolytic activity of conditioned medium of TZD treated pancreatic cancer cell. Cells were starved in serum free medium for 24 hours, washed, and then cultured in serum free medium with 20 μM of TZD (RGZ or PGZ) or their vehicle for further 24 hours. Supernatants were then collected and concentrated. Aliquots of culture medium (5 μg of proteins) were subjected to sodium dodecyl sulfate-polyacrylamide slab gel electrophoresis and then transferred onto nitrocellulose filter as described in Methods. Plasminogen activator activity corresponding band (54 kDa) was significantly reduced in conditioned medium of both PPARγ expression and non-expressing cells. A representative of three independent experiments performed in duplicate, yielding similar results, is shown.
DISCUSSION
We have studied the effects of two TZD, pioglitazone (PGZ) and rosiglitazone (RGZ), on pancreatic cancer cell growth and invasiveness, and have characterised the relation between PPARγ expression and their anticancer properties. Firstly, we analysed the expression of PPARγ in a series of 42 pancreatic cancers and in 10 pancreatic cancer cell lines. RT-PCR analysis showed that only about 55% of human cancer expressed the receptor and this does not seem to be correlated with either the histological grade or the stage (table 1 ▶ and fig 1 ▶). No PPARγ expression could be also detected in three cell lines (HPAC, PL45, and SU86.86) (fig 2 ▶).
The two TZD similarly induced a strong inhibition of the ability of pancreatic cancer cells to migrate through Matrigel coated invasion membranes. Surprisingly, after the TZD treatment we found a similar dose dependent inhibition of cell invasiveness in both PPARγ expressing and non-expressing cell lines (figs 4 ▶ and 5 ▶). On the contrary both RGZ and PGZ inhibited growth in PPARγ expressing cells but had no effect in the PPARγ deficient cells (figs 6 ▶ and 7 ▶). The PPARγ independent mechanism in TZD inhibition of pancreatic cells invasiveness was confirmed by using specific PPARγ antagonists. Both BADGE and GW9662 completely blocked the TZD induced growth arrest but had no effect on invasiveness (fig 8 ▶).
These data are partially in contrast with previous observation regarding the PPARγ independent anticancer effect of TZD. In fact, in PPARγ-/- embryonic stem cells, troglitazone induces growth arrest by inhibition of translation initiation with partial depletion of intracellular Ca2+ stores and activation of the double stranded RNA dependent protein kinase (PKR).31 Furthermore, Abe et al have indicated that troglitazone suppressed cell growth and histamine secretion by a PPARγ independent mechanism in the human basophilic leukaemia cell line KU812.36 This may be explained by the different chemical structure of the various members of the TZD family. Troglitazone, for instance, has the chroman structure of vitamin E, suggesting that this TZD could regulate signal pathways by mimicking the effects of vitamin E, and independently of PPARγ transcriptional activation.37
Proteolytic matrix degradation is a mandatory event in tumour invasion and metastasis.38 Among the several classes of proteinases involved in the invasive potential of pancreatic cancer the gelatinases and the plasminogen activator system play a primary role.35,39,40 Supernatants of both PPARγ expressing and non-expressing cell cultures treated with TZD showed a drastic inhibition of the MMP-2 gelatinolytic activity (fig 9 ▶) and this effect was correlated with a significant inhibition of MMP-2 gene expression (fig 11A and 11B ▶ ▶). The effect of TZD on MMP-2 expression are particularly relevant in consideration of the evidence indicating that inhibition of MMP-2 seems to be most important in prolonging survival in a murine model of pancreatic cancer 41,42 and that MMP-2 has a primary role in pancreatic cancer cell invasion.35 Furthermore, it has been shown that the MMP-2 activation ratio is also significantly higher in pancreatic carcinoma specimens with histologically positive regional lymph node metastasis and distant metastasis than those without metastasis.43
Several in vivo and in vitro studies have also clearly shown an important role of the fibrinolytic system in epithelial tumour progression.44,45 Overexpression of constituents of the plasminogen activator system, such as uPA and uPAR, were shown in pancreatic cancer.40 We have shown that TZD treatment drastically inhibits fibrinoyitic activity in both PPARγ expressing and non-expressing cells (fig 10 ▶) and that this effect was correlated with a dramatic upregulation of plasminogen activator inhibitor PAI-1, the major physiological inhibitor of fibrinolysis (fig 11A and 11B ▶ ▶).
Previous studies have shown the ability of TZD to inhibit migration of smooth muscle and endothelial cells, but the correlation with PPARγ activation was not investigated.46,47 In smooth muscle cells, TZD inhibit growth factor induced migration by upregulation of PTEN (a phosphatase that functions as a negative regulator of PI3K/Akt signalling) and inhibit the expression of transcription factor Ets-1,48 which is required for matrix metalloproteinase induction and tissue invasion by vascular smooth muscle cells.49 Similarly, in bovine endothelial cells TZD inhibit vascular endothelial growth factor induced migration and suppress its production, and inhibit MMP activity.50–52 In addition, studies in human endothelial cells have shown that PPARγ ligands increase PAI-1 gene expression and inhibit angiogenesis both in vivo and in vitro.51–53 The PPARγ independent mechanisms by which TZD control MMP-2 and PAI-1 gene expression remain speculative. Chawla et al reported that PPARγ is required for the positive effects of its ligands in modulating macrophage lipid metabolism, but that the inhibitory effects on cytokine production and gelatinase gene expression may be PPARγ independent.54 Interestingly, a rapid non-genomic action of TZD was shown in smooth muscle cells, where PGZ activates extracellular signal regulated protein kinase (ERK) and leads to c-fos mRNA induction within 30 minutes via a phosphatidyl inositol 3 (PI3)-kinase dependent mechanism.55 It has been shown that PI3-kinase activation is involved in PAI-1 regulated expression.56 Thus, it might also be involved in the rapid TZD mediated induction of PAI-1 gene expression in pancreatic cancer cells. Preliminary results demonstrated that wortmannin, a PI3K inhibitor, partially reduced the effect of TZD on the ability of pancreatic cancer cells to invade Matrigel and abrogated the stimulated expression of PAI-1, but had negligible effect on MMP-2, suggesting different non-genomic effects of these drugs (Galli A et al, unpublished observation, September 2003).
In conclusion, it is tempting to postulate that TZD have a dual pharmacological effect on pancreatic cancer cells. On one hand they inhibit cell growth and induce ductal differentiation by PPARγ transactivation and transcription of an array of PPARγ responsive genes; on the other hand TZD inhibit cell invasiveness by inhibition of gelatinolytic and fibrinolytic activity by way of a mechanism totally independent of PPARγ expression and activation. In view of our new findings and in consideration that PGZ and RGZ are already orally administered, FDA approved drugs which have been used in over two million people with minimal side effects, they hold great promise to complement conventional chemotherapeutic regimens for pancreatic cancer therapy.
Acknowledgments
This work was supported by grants from Ministero dell’Università, della Ricerca Scientifica e Tecnologica, (MURST), Italian Society of Gastroenterology (SIGE), and BYK Gulden Italia.
We are indebted to Dr A Scarpa for his gift of PK-1 cell line and to Dr M Breyer for human PPARγ cDNA probe. The authors express their appreciation to Professor M Serio and Professor M Mannelli for many helpful comments and suggestion.
Abbreviations
BADGE, bisphenol A diglycidyl ether
DMEM, Dulbecco’s modified Eagle’s medium
FBS, fetal bovine serum
GAPDH, glyceraldehyde phosphatase dehydrogenase
MMP-2, metalloproteinase-2
PAI-1, plasminogen activator inhibitor 1
PGZ, pioglitazone
PPAR, peroxisome proliferators activated receptor
PPRE, peroxisome proliferator response element
RGZ, rosiglitazone
RPA, RNase protection assay
RT-PCR, reverse transcriptase polymerase chain reaction
TZD, thiazolidinediones
REFERENCES
- 1.Silverberg E , Boring CC, Squires TS. Cancer statistics 1990. Can Cancer J Clinitians 1990;40:9–26. [PubMed] [Google Scholar]
- 2.Hohne MW, Halatsch ME, Kahl GF, et al. Frequent loss of expression of the potential tumor suppressor gene DCC in ductal pancreatic adenocarcinoma. Cancer Res 1992;52:2616–19. [PubMed] [Google Scholar]
- 3.Bakkevold KE, Arnesjo B, Kambestang B. Carcinoma of the pancreas and Papilla of Vater: presenting symptoms, signs, and diagnosis related to stage and tumor site. A prospective multicenter trial in 472 patients. Scand J Gastroenterol 1992;27:317–25. [DOI] [PubMed] [Google Scholar]
- 4.Rocha Lima CM, Centeno B. Update on pancreatic cancer. Curr Opin Oncol 2002;14:424–30. [DOI] [PubMed] [Google Scholar]
- 5.Olefsky JM. Treatment of insulin resistance with peroxisome-proliferator-activated receptor γ agonists. J Clin Invest 2000;106:467–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braissant O , Foufelle F, Scotto M, et al. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR alpha, beta and gamma in adult rats. Endocrinology 1996;137:354–66. [DOI] [PubMed] [Google Scholar]
- 7.Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA 1994;91:7355–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts-Thomson SJ. Peroxisome proliferator-activated receptors in tumorigenesis: targets of tumor promotion and treatment. Immunol Cell Biol 2000;78:436–41. [DOI] [PubMed] [Google Scholar]
- 9.Pignatelli M , Cortes-Cantelli M, Lai C, et al. The peroxisome proliferator-activated receptor γ is an inhibitor of ErbBs activity in human breast cancer cell. J Cell Sci 2001;114:4117–26. [DOI] [PubMed] [Google Scholar]
- 10.Brockman JA, Gupta RA, Du Bois RN. Activation of PPARgamma leads to inhibition of anchorage independent growth of human colorectal cancer. Gastroenterology 1998;115:1049–55. [DOI] [PubMed] [Google Scholar]
- 11.Sarraf P , Mueller E, Jones D, et al. Differentiation and reversal of malignant changes in colon cancer through PPARγ. Nat Med 1998;4:1046–52. [DOI] [PubMed] [Google Scholar]
- 12.Kroll TG, Sarraf P, Pecciarini L, et al. PAX8-PPARγ1 fusion oncogene in human thyroid carcinoma. Science 2000;289:1357–60. [DOI] [PubMed] [Google Scholar]
- 13.Sarraf P , Mueller E, Smith WM, et al. Loss-of-function mutations in PPAR gamma associated with human colon cancer. Mol Cell 1999;3:799–804. [DOI] [PubMed] [Google Scholar]
- 14.Motomura W , Okumura T, Takahashi N, et al. Activation of peroxisome proliferator-activated receptor γ by troglitazone inhibits cells growth through the increase of p27 in human pancreatic carcinoma cell. Cancer Res 2000;60:5558–64. [PubMed] [Google Scholar]
- 15.Kawa S , Nikaido T, Unno H, et al. Growth inhibition and differentiation of pancreatic cancer cell lines by PPARγ ligand troglitazone. Pancreas 2002;1:1–7. [DOI] [PubMed] [Google Scholar]
- 16.Albini A , Iwamoto Y, Kleinman HK. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res 1987;47:3239–45. [PubMed] [Google Scholar]
- 17.Laemli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–2. [DOI] [PubMed] [Google Scholar]
- 18.Andrews NC, Faller DV. A rapid micropreparation technique for extraction of DNA-binding proteins from limiting numbers of mammalian cells. Nucl Acids Res 1991;19:2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chomczynski P , Sacchi E. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987;162:156–9. [DOI] [PubMed] [Google Scholar]
- 20.Casini A , Pinzani M, Milani S, et al. Regulation of extracellular matrix synthesis by transforming growth factor β1 in human fat-storing cells. Gastroenterology 1993;105:245–53. [DOI] [PubMed] [Google Scholar]
- 21.Davis EG, Pintar Allen KA, Salazar R, et al. Matrix metalloproteinase-1 and-9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J Cell Sci 2000;114:917–30. [DOI] [PubMed] [Google Scholar]
- 22.Guan Y , Zhang Y, Davis L, et al. Expression of peroxisome proliferator-activated receptors in urinary tract of rabbits and humans. Am J Physiol 1997;273 (Renal Physiol 42) :F1013–F1022. [DOI] [PubMed] [Google Scholar]
- 23.Basset P , Bellocq JP, Wolf C, et al. A novel metalloproteinase gene specifically expressed in stromal cells of breast carcinoma. Nature 1990;348:699–704. [DOI] [PubMed] [Google Scholar]
- 24.Herbst H , Wege T, Milani S, et al. Tissue inhibitor of metalloproteinases-1 and 2 RNA expression in rat and human liver fibrosis. Am J Pathol 1997;150:1647–59. [PMC free article] [PubMed] [Google Scholar]
- 25.Huhtala P , Chow LT, Tryggvason K. Structure of human typeIV collagenase gene. J Biol Chem 1990;265:11077–82. [PubMed] [Google Scholar]
- 26.Fibbi G , Magnelli L, Pucci M, et al. Interaction of urokinase A chain with the receptor of human stimulates release of urokinase-like plasminogen activator. Exp Cell Res 1990;187:33–8. [DOI] [PubMed] [Google Scholar]
- 27.Galli A , Stewart M, Dorris, et al. High-level expression of RXRα and the presence of endogenous ligands contribute to expression of a peroxisome proliferator-activated receptor-responsive gene in hepatoma cells. Arch Biochem Biophys 1998;354:288–98. [DOI] [PubMed] [Google Scholar]
- 28.de Wet JR, Wood KV, DeLuca M, et al. Firefly luciferase gene: structure and expression in mammalian cells. Mol Cell Biol 1987;7:725–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crabb DW, Dixon JE. A method for increasing the sensitivity of chloramphenicol acetyltransferase assay in extracts of transfected cultured cells. Anal Biochem 1987;163:88–92. [DOI] [PubMed] [Google Scholar]
- 30.Quattrone A , Fibbi G, Anichini E, et al. Reverse of invasive phenotype of transformed human fibroblast by anti-messenger oligonucleotide inhibition of urokinase receptor gene expression. Cancer Res 1995;55:90–5. [PubMed] [Google Scholar]
- 31.Palakurthi SS, Aktas H, Grubissich LM, et al. Anticancer effects of thiazolidinediones are independent of peroxisome proliferatror-activated receptor γ and mediated by inhibition of translation initiation. Cancer Res 2001;61:6213–18. [PubMed] [Google Scholar]
- 32.Wright HM, Clish CB, Mikami T, et al. A synthetic antagonist for the peroxisome proliferator-activated receptor γ inhibits adipocyte differentiation. J Biol Chem 2000;275:1873–7. [DOI] [PubMed] [Google Scholar]
- 33.Clay CE, Monjazeb A, Thorburn J, et al. 15–deoxy Δ12,14-prostaglandin J2-induced apoptosis does not require PPARγ in breast cancer cell. J Lipid Res 2002;43:1818–28. [DOI] [PubMed] [Google Scholar]
- 34.Bramhall SR, Neoptolemos JP, Stamp GW, et al. Imbalance of expression of matrix metalloproteinases (MMPs) and tissue inhibitors of the matrix metalloproteinases (TIMPs) in human pancreatic carcinoma. J Pathol 1997;182:347–55. [DOI] [PubMed] [Google Scholar]
- 35.Ellendrieder V , Hendler SF, Ruhland C, et al. TGF-β-induced invasiveness of pancreatic cancer cells is mediated by matrix metalloprotease-2 and the urokinase plasminogen activator system. Int J Cancer 2001;93:204–11. [DOI] [PubMed] [Google Scholar]
- 36.Abe A , Kiriyama Y, Hirano M, et al. Troglitazone suppresses cell growth of KU812 cells independently of PPARγ. Eur J Pharmacol 2002;436:7–13. [DOI] [PubMed] [Google Scholar]
- 37.Inoue I , Katayama S, Takahashi K, et al. Troglitazone has a scavenging effect on reactive oxygen species. Biochem Biophys Res Commun 1997;235:113–16. [DOI] [PubMed] [Google Scholar]
- 38.Brinckerhoff CE, Rutter JL, Benbow U. Interstitial collagenases as markers of tumor progression. Clin Cancer Res 2000;6:4823–30. [PubMed] [Google Scholar]
- 39.Ellendrieder V , Alber B, Lacher U, et al. The role of MT-MMPs and MMP-2 in the progression of pancreatic cancer. Int J Cancer 2000;85:14–20. [DOI] [PubMed] [Google Scholar]
- 40.Cantero D , Friess H, Deflotrin J, et al. Enhanced expression of urokinase plasminogen activator and its receptor in pancreatic carcinoma. Br J Cancer 1997;75:388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zervos E , Shafii A, Rosemurgy AS. Matrix metalloproteases (MMP) inhibition selectively decreases type II MMP in a murine model of pancreatic cancer. J Surg Res 1999;81:65–68. [DOI] [PubMed] [Google Scholar]
- 42.Haq M , Shafii A, Zervos E, et al. Addition of matrix metalloproteinase inhibition to conventional cytotoxic therapy reduces tumor implantation and prolongs survival in a murine model of human pancreatic cancer. Cancer Res 2000;60:3207–11. [PubMed] [Google Scholar]
- 43.Koshiba T , Hosotani R, Wada M, et al. Involvement of matrix metalloproteases-2 activity in invasion and metastasis of pancreatic carcinoma. Cancer 1998;82:642–50. [DOI] [PubMed] [Google Scholar]
- 44.Mohanam S , Chintala SK, Go Y, et al. In vitro inhibition of human glioblastoma cell lines invasiveness by antisense uPA receptor. Oncogene 1997;14:1351–9. [DOI] [PubMed] [Google Scholar]
- 45.Foekens JA, Peters HA, Look MP, et al. The urokinase system of plasminogen activation and prognosis in 2780 breast cancer patients. Cancer Res 2000;60:636–43. [PubMed] [Google Scholar]
- 46.Hsueh WA, Law RE. PPARγ and atherosclerosis: effects on cell growth and movement. Arterioscler Thromb Vasc Biol 2001;21:1891–5. [DOI] [PubMed] [Google Scholar]
- 47.Xin X , Yang S, Kowalski J, et al. Peroxisome proliferator-activated receptor γ ligands are potent inhibitors of angiogenesis in vitro and in vivo. J Biol Chem 1999;274:9116–21. [DOI] [PubMed] [Google Scholar]
- 48.Goetze S , Bungenstock A, Czupalla C, et al. Leptin induces endothelial cells migration through Akt, which is inhibited by PPAR-γ ligands. Hypertension 2002;40:748–54. [DOI] [PubMed] [Google Scholar]
- 49.Sato Y . Transcription factor Ets-1 as a molecular target for angiogenesis. Hum Cell 1998;11:207–14. [PubMed] [Google Scholar]
- 50.Goetze S , Eilers F, Bungenstock A, et al. PPAR activators inhibit endothelial cell migration by targeting Akt. Biochem Biophys res Commun 2002;293:1431–7. [DOI] [PubMed] [Google Scholar]
- 51.Xin X , Yang S, Kowalsky J, et al. Peroxisome proliferator-activated receptor γ ligands are potent inhibitors of angiogenesis in vitro and in vivo. J Biol Chem 1999;274:116–21. [DOI] [PubMed] [Google Scholar]
- 52.Pinigrahy D , Singer S, Shen LQ, et al. PPARγ ligands inhibit primary tumor growth and metastasis by inhibiting angiogenesis. J Clin Invest 2002;110:923–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marx N , Bouncier T, Sukhova GK, et al. PPARγ activation in human endothelial cells increases plasminogen activator inhibitor type-1 expression. Arterioscler Thromb Vasc Biol 1999;19:546–51. [DOI] [PubMed] [Google Scholar]
- 54.Chawla A , Barak Y, Nagy L, et al. PPARγ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 2001;7:48–52. [DOI] [PubMed] [Google Scholar]
- 55.Takeda K , Ichiki T, Tokonou T, et al. A. 15-deoxyprostaglandin J2 and thiazolidinediones activate the MEK/ERK pathway through phospatidylinositol 3-kinase in vascular smooth muscle cells. J Biol Chem 2001;276:48950–5. [DOI] [PubMed] [Google Scholar]
- 56.Kietzmann T , samoylenko A, Roth U, et al. Hypoxia-inducible factor-1 and hypoxia response elements mediate the induction of plasminogen activator inhibitor-1 gene expression by insulin in primary rat hepatocytes. Blood 2003;101:907–14. [DOI] [PubMed] [Google Scholar]