

SUMMARY
The majority of pancreatic diseases are associated with genetic polymorphisms. Recent breakthroughs in understanding the origin and pathways toward pancreatic diseases, and especially acute and chronic pancreatitis, reveal that specific variation in the genomic DNA sequence of individuals strongly influence their susceptibility to pancreatitis, the severity and nature of the inflammatory process, and the likelihood of various complications. Acute pancreatitis is an event, and chronic pancreatitis is a process. They are sequentially linked with chronic pancreatitis reflecting a pathophysiological response to acute pancreatitis events. The triggers, thresholds, mechanism of injury, and immunological responses of individuals with acute and chronic pancreatitis are being organised and defined. In the future, early use of genetic testing will likely play a critical role in early diagnosis and prognosis of pancreatic diseases, and could guide new and effective preventative and therapeutic interventions. The benefits and risk of limited genetic testing have been defined by consensus conference, while the framework for future and more extensive genetic evaluation is still developing.
INTRODUCTION
Diseases of the pancreas continue to be among the most mysterious and difficult of all human diseases to study. The pancreas is inaccessible to physical examination because of its retroperitoneal location, and remains invisible to standard radiographic examination because it lacks bony structure. Biopsies are also rarely done for fear of triggering acute pancreatitis, creating fistula, or initiating bleeding. These factors limited our understanding of pancreatic physiology and pathophysiology, limited early diagnosis, and precluded development of effective treatment for all major pancreatic diseases, until now.
Growing evidence suggests that most diseases of the pancreas have genetic predispositions and/or basis. This should not be surprising as the pancreas is an organ shielded from direct exposure to a hostile environment (unlike the skin, lungs, and digestive tract), and neither filters nor detoxifies most noxious agents (unlike the kidney and liver). As a result, environmental factors appeared to play a limited role in pancreatic diseases. Thus understanding pancreatic disease has lagged behind advances in other diseases until new genetic methods became available. The latest evidence demonstrates that idiopathic and alcoholic acute pancreatitis, and especially the various forms of chronic pancreatitis, are most often complex disorders that occur in individuals having multiple risk factors for susceptibility and severity that together contribute to the aetiology through pathological pathways that are activated by triggering factors. Understanding these interactions will provide the rationale for future evaluation and treatment.
We are now on the verge of a new era in testing and treating diseases of the pancreas. But to understand the timing, purpose, and value of genetic testing, one must understand the clinical and pathophysiological context. This includes the origin, aetiologies, and processes leading to acute and chronic pancreatitis, and the genetic alterations known to contribute to these disorders. This framework should also be useful for categorising and applying new mutations to our understanding of pancreatic diseases. The major issues associated with genetic testing will be discussed from this context. The current knowledge provides genetic insight into acute pancreatitis, with the understanding that recurrent pancreatitis, in some cases, leads to chronic pancreatitis. In our view, chronic pancreatitis requires three “hits”, which can include environmental stressors, pancreatic injury from trypsinogen activation, and an inappropriate immunological response leading to chronic inflammation and/or fibrosis. Specific genetic testing in the future should predict who will have recurrent acute pancreatitis and who will progress to chronic pancreatitis.
Genetic testing is one of the most important and powerful tools ever developed. When properly performed, results are 100% sensitive and specific for the detection of specific DNA sequence variations, and the results remain valid for a lifetime. However, diseases with genetic risk are often complex so that a variety of different mutations in the same gene must be considered (for example, allelic heterogeneity), multiple genes must be considered for polygenic disorders, and only a proportion of individuals with specific mutation may develop disease (for example, incomplete penetrance).1 The reason that these tests are so powerful is not in accuracy, but in the implications, both negative and positive. Unfortunately, the negative aspects tend to be recognised early because of concerns of disease risk for family members, impact on family relationships, career choices, employment, insurability, and other forms of discrimination.2–4 The positive implications are for long term health through therapeutics or lifestyle modification, although this may take some time to demonstrate in chronic progressive diseases with significant heterogeneity.
In this review, the focus will be on three genes that are associated with acute and recurrent acute pancreatitis, and that lead to chronic pancreatitis: trypsinogen, SPINK1, and CFTR. Other genes associated with chronic pancreatitis will also be considered. Genetic testing is not yet available or recommended for most of these genes but this situation will likely change with continued research.
TRYPSIN AND ACUTE PANCREATITIS
In simple terms, acute pancreatitis is caused by unregulated trypsin activity within the pancreatic acinar cell or pancreatic duct that leads to zymogen activation, pancreatic autodigestion, and pancreatic inflammation (that is, pancreatitis).5,6 The primary factors that trigger an episode of acute pancreatitis include pancreatic hyperstimulation, biliary disease, and alcohol. Hyperstimulation of the pancreas causes a sustained rise in intra-acinar cell calcium levels, especially within the apical region where zymogen granules are stored.7,8 If trypsinogen is activated to trypsin in the context of intracellular hypercalcaemia, then trypsin activity is sustained and pancreatitis is initiated. Recent studies also demonstrated that the pancreatic acinar cells are exquisitely sensitive to bile acids.9 Exposure of the luminal surface of the acinar cell to bile acids causes a marked increase in intracellular calcium, and therefore may also cause acute pancreatitis through a calcium dependent mechanism, as described below.
Trypsin is a major pancreatic protease, targeting peptide chains at the C terminal side of interior arginine (R) or lysine (K) residues. Structurally, trypsin is a typical serine protease with two globular protein domains connected by a single side chain on the surface opposite the active site (fig 1 ▶, autolysis loop). Trypsinogen becomes trypsin with cleavage of a short exposed peptide chain called trypsinogen activation peptide by the action of enterokinase or by a second trypsin molecule. The trypsin molecule contains a calcium binding pocket (fig 1 ▶, calcium) near the side chain connecting the two globular domains of the molecule. This side chain (also called the autolysis loop) contains an arginine residue at amino acid position 117 (coded for by codon 122—that is, R122), which is a target for attack by other trypsin enzymes. Enzymatic cleavage of the side chain at R122 by the second trypsin leads to rapid destruction of the first trypsin molecule (autolysis). The autolysis loop is flexible and R122 may come close to the calcium binding pocket. As the concentration of soluble calcium rises, calcium enters the calcium binding pocket and limits exposure of R122 to enzymatic attack by another trypsin.10 The importance of calcium in protecting trypsin from autolysis has been demonstrated in vitro where trypsin rapidly destroys itself, unless calcium is added.11 The role of calcium has also been demonstrated in vivo. In rats, hypercalcaemia causes pancreatitis only after pancreatic stimulation (which opens calcium channels to allow calcium to flood into the pancreatic acinar cells) and after trypsinogen activation to trypsin,12 which is presumably stabilised by calcium. Taken together, trypsin is susceptible to rapid autolysis within the acinar cell where calcium levels are (usually) low, protected from autolysis after active secretion into the pancreatic duct and duodenum where calcium levels are high, and then undergoes autolysis in the distal small intestine after calcium is absorbed in the distal duodenum and jejunum. Thus calcium appears to play a central role in trypsin secretion, trypsin stabilisation, and some forms of pancreatitis (see review by Sutton and colleagues13).
Figure 1.

Structural features of trypsin (PRSS1) and PSTI (SPINK1). Residues in cationic trypsin (PRSS1 gene–blue and yellow) include the active catalytic site (Ac), R122, and V118 within the autolysis loop, the calcium binding loop with calcium (pink), and the residue interacting with V118 of the autolysis loop. N29 is shown near the activation site. In the PSTI (SPINK1 gene–Red), N34 and P55 are shown and residue entering the specificity pocket of trypsin (SP). N29 and R122 of PRSS1 are commonly mutated in hereditary pancreatitis (illustration provided by Andrew Brunskill PhD).
Acute pancreatitis develops with unregulated trypsin activity after breakdown of the critical protective mechanisms. Intracellular protective mechanisms include synthesis of trypsin as an inactive zymogen (trypsinogen), zymogens compartmentalisation and packaging, synthesis of a specific trypsin inhibitor (pancreatic secretory trypsin inhibitor, PSTI or SPINK1), R122 dependent trypsinogen/trypsin autolysis, control of intra-acinar cell calcium levels (to facilitate autolysis), and lysosome dependent pathways of zymogen/activated digestive enzyme elimination. Once the zymogens are secreted into a calcium rich juice (which eliminates the autolysis protective mechanism) then the pancreas is dependent on SPINK1 to inhibit prematurely activated trypsinogen, and rapid flushing of the pancreatic duct by fluid from the duct cells. Duct flushing is a cystic fibrosis transmembrane conductance regulator (CFTR) dependent protective mechanism. Disruption of any of these protective mechanisms increases susceptibility to acute pancreatitis and predisposes to chronic pancreatitis. The combined effect of environmental and genetic factors in increasing the risk of recurrent acute pancreatitis is given in fig 2 ▶.
Figure 2.

Susceptibility to acute pancreatitis (AP). Hypothetical relationship between pancreatitis risk factors and developing episodes of acute pancreatitis. Two subjects are illustrated who have identical environmental exposures but few genetic risk factors (low risk, bottom line) or significant risk factors (high risk, top line). The risk of developing pancreatitis at any point in time is likely variable, and reflects the combination of susceptibility factors and triggering factors that alter risk (for example, meals, alcohol, biliary, etc). Acute pancreatitis develops when the protective mechanisms are no longer adequate and the combined genetic and environmental risk factors and environmental triggers drive the pancreas over the threshold needed to initiate acute pancreatitis (AP threshold, broken line). Note that the subject with the higher underlying risk develops clinically recognised acute pancreatitis after several triggering events whereas the low risk subject does not develop acute pancreatitis with the same events.
GENETIC TESTING IN RECURRENT ACUTE PANCREATITIS
In most cases, acute pancreatitis is caused by gall stones or alcohol and no genetic testing is indicated. Unexplained recurrent acute pancreatitis however may be associated with known genetic mutations in the cationic trypsinogen gene (PRSS1), the SPINK1 gene, or the CFTR gene. Other genes are also likely important but they are yet to be discovered and understood within the context of pancreatic disease. The age of symptom onset and family history are clues to the likelihood of finding an underlying susceptibility gene mutation. Currently, the only gene for which genetic testing is recommended is trypsinogen.14–16
Trypsinogen mutations
The importance of trypsin was discovered in studies of hereditary pancreatitis, an unusual form of acute and chronic pancreatitis that runs in families following an autosomal dominant inheritance pattern. Most cases of hereditary pancreatitis are associated with mutations in the cationic trypsinogen gene (PRSS1).17,18 Although nearly 20 pancreatitis associated mutations in the PRSS1 gene have been described, the R122H and N29I mutations (also numbered as R117H and N21I using the chymotrypsinogen amino acid numbering system) represent the vast majority of cases. These are gain of function mutations that likely interfere with autolysis and/or cause premature trypsinogen activation.19,20 In families with PRSS1 R122H or N29I mutations, between 60% and 80% of individuals who inherit the mutation usually develop pancreatitis.21–23 In addition, approximately half of individuals with acute pancreatitis will develop chronic pancreatitis, and up to 40% of individuals with chronic pancreatitis will develop pancreatic cancer.24,25 In most cases, a patient with a gain of function PRSS1 gene will identify at least one other family member with recurrent acute or chronic pancreatitis.18 In other cases, no family history is reported either because the history is unknown, the diagnosis of pancreatitis was not made or recorded by family members in previous generations, or the patient has few relatives. However, pancreatitis from PRSS1 mutations is uncommon, usually represents 10% or less of most clinical cohorts with chronic pancreatitis.18
The different clinical courses of pancreatitis in individuals with hereditary pancreatitis appear to involve both environmental and genetic modifiers (unpublished observations) while the incomplete disease penetrance is complex26 and likely reflects the effects of environmental triggers. Even identical twins with PRSS1 mutations can be discordant for affected status (thus arguing for environmental triggers) but similar in clinical course when both are affected (arguing for modifier genes).26 Thus the major susceptibility factor for hereditary pancreatitis is known but the major disease modifying factors are yet to be identified.
Very accurate diagnostic tests are now available for the major trypsinogen gene mutations. Genetic diagnosis of hereditary pancreatitis has significant implications for patients and their extended family3 whereas diagnosis of other types of pancreatitis does not have the same impact. Therefore, the diagnosis of hereditary pancreatitis should be specifically limited to patients with gain of function PRSS1 mutations27 or otherwise unexplained pancreatitis in an individual from a family in which the pancreatitis phenotype appears to be inherited through a disease causing gene mutation expressed in an autosomal dominant pattern.28,29Familial pancreatitis refers to pancreatitis from any cause that occurs in a family with an incidence that is greater than would be expected by chance alone, given the size of the family and incidence of pancreatitis within a defined population.28 This distinction is important from prognostic, genetic counselling, and family planning perspectives, and because of potential of insurance discrimination associated with a “genetic disease”.2,3
The role of genetic testing in hereditary pancreatitis has been addressed in several recent reviews,2,3 a consensus conference report,14 and recommendations are listed on the National Guideline Clearing House website (www.guideline.gov) under “Genetic testing for hereditary pancreatitis: guidelines for indications, counselling, consent and privacy issues”. PRSS1 genetic testing is recommended in symptomatic patients with any of the following: (1) recurrent (two or more separate documented episodes of typical pain with hyper-amylasaemia) attacks of acute pancreatitis for which there is no explanation (for example, anatomical anomalies, ampullary or main pancreatic strictures, trauma, viral infection, gall stones, alcohol, drugs, hyperlipidaemia, etc.), (2) unexplained (idiopathic) chronic pancreatitis, (3) a family history of pancreatitis in a first degree (parent, sib, child) or second degree (aunt, uncle, grandparent) relative, (4) an unexplained episode of documented pancreatitis occurring in a child that has required hospitalisation, and where there is significant concern that hereditary pancreatitis should be excluded, or (5) as part of an approved research protocol.14
Genetic testing in children represents an additional set of issues, especially surrounding informed consent. Under age 12 years the child usually cannot reasonably participate in a decision making process of whether or not to undergo genetic testing, and only partially enters into this process until age 16 years. Thus the consensus committed suggested that genetic testing of a child (under the age of 16 years) for PRSS1 mutations should be considered after (1) an episode of documented pancreatitis of unknown aetiology and severe enough to require hospitalisation, (2) two or more documented episodes of pancreatitis of unknown aetiology, (3) an episode of documented pancreatitis occurring in a child where a relative is known to carry an hereditary pancreatitis mutation, (4) a child with recurrent abdominal pain of unknown aetiology where the diagnosis of hereditary pancreatitis is a distinct clinical possibility, or (5) chronic pancreatitis of unknown aetiology where the diagnosis of hereditary pancreatitis is a distinct clinical possibility.14
The issue of genetic counselling goes hand in hand with genetic testing.14 For PRSS1 testing, the ordering physician should explain why the test is being ordered, autosomal dominant mode of inheritance, incomplete penetrance, and variable clinical course. If a major PRSS1 mutation is identified (for example, R122H or N29I) there is a significant lifetime risk of pancreatic cancer in patients with longstanding chronic pancreatitis,24 and this risk is lower in individuals avoiding tobacco smoking.30 In addition, patients’ relatives may be at risk, and there should be a plan for providing counselling and possibly genetic testing for family members if warranted. As there is ongoing research for reducing the risk, frequency, and severity of hereditary pancreatitis, patients should be informed of possible research study participation opportunities.
FUTURE OF GENETIC TESTING IN RECURRENT ACUTE PANCREATITIS
Growing evidence suggests that a significant fraction of cases of idiopathic recurrent acute pancreatitis and idiopathic chronic pancreatitis have strong genetic components. Mutations in several genes, including the CFTR and SPINK1 genes, have been repeatedly associated with idiopathic chronic pancreatitis in children,31 families,32 adults,33,34 and alcoholics.33,34 These mutations usually result in proteins with a loss of function, but unlike PRSS1 mutations, most individuals (>99%) with heterozygous mutations in either of these pancreatitis associated gene do not develop acute or chronic pancreatitis.15,16,35 However, there appears to be an association between heterozygous mutations in the SPINK1 and CFTR genes and unexplained pancreatitis in various patient populations.32,36 Furthermore, heterozygous SPINK1 or CFTR mutations appear more commonly in idiopathic pancreatitis than homozygous or compound heterozygous mutations in these genes, as predicted for autosomal recessive diseases.32 Finally, heterozygous SPINK1 and CFTR gene mutations are often enriched in familial pancreatitis families that may not follow classic Mendelian inheritance patterns. This suggests that these mutations are components of complex genetic traits. Genetic testing will become important as the critical factors and interactions are identified and interpretation and prediction become possible.
SPINK1 mutations
As noted above, the pancreas synthesises a specific trypsin inhibitor, SPINK1. Between 1% and 4% of the world’s population appear to have a complex allele (N34S, with IVS1-37T>C, IVS2+286 A>G, IVS3-604 G>A, and IVS3-66-65insTTTT).35 As these polymorphisms appear to be tightly linked (inherited together based on close physical proximity), N34S is usually the only polymorphism investigated. Witt and colleagues31 were the first to demonstrate an association between SPINK1 polymorphisms and idiopathic chronic pancreatitis in children even though it had been previously excluded as a hereditary pancreatitis gene by Chen and colleagues37 and was noted to be a common polymorphism in the French population.37 Although the SPINK1 N34S allele is associated with multiple types of acute and chronic pancreatitis,38 the association is weak, with <1% of mutation carriers developing pancreatitis sometime during their lifetime.32,35 Furthermore, the severity of pancreatitis appears to be similar between subjects with heterozygous, homozygous, or compound heterozygous genotypes that include the N34S allele32 suggesting that the genetics is complex. Therefore, we hypothesised that the SPINK1 N34S allele was acting as either a modifier gene or a susceptibility factor for a polygenic complex trait.32,36,38,39 In addition, other rare SPINK1 polymorphisms have been identified that are associated with a higher risk of pancreatitis than the N34S genotype.31
Recently, Felderbauer and colleagues40 identified an interesting interaction between SPINK1 and mutations in the calcium sensing receptor gene. These mutations cause familial hypocalciuric hypercalcaemia, an autosomal dominant disorder with nearly 100% penetrance characterised by benign elevation in plasma calcium levels.40 Patients with both HFF and the SPINK1 N34S allele mutations had dozens of episodes of recurrent abdominal pain, pancreatitis, and eventually developed chronic pancreatitis. Thus the SPINK1 N34S allele modifies the HFF phenotype to cause recurrent acute and chronic pancreatitis, just as it modifies the phenotypic expression of mild CFTR mutations to cause atypical cystic fibrosis associated pancreatitis.41 If SPINK1 N34S is acting as a disease modifier, heterozygous SPINK1 N34S should only be associated with patients in the presence of other gene mutations. This hypothesis has yet to be fully explored and tested.
CFTR mutations
The CFTR anion channel is a critical molecule for correct functioning of pancreatic duct cells and other anion secreting epithelial cells. CFTR conducts both chloride and bicarbonate, and the opening and closing of this channel controls the bulk of pancreatic fluid secretion. CFTR is a master channel that also regulates the function of other channels. The strong association between CFTR mutations and idiopathic chronic pancreatitis was demonstrated in 1998,33,34 and the role of CFTR gene mutations in alcoholic chronic pancreatitis has been investigated.42 However, in common with the study of SPINK1 mutations, our understanding of the genetic risk of CFTR mutations for pancreatitis remains limited. Interpretation of the importance of CFTR mutations in chronic pancreatitis suffers from the following issues: the CFTR gene is large and very expensive for most clinical investigators to sequence; and the major mutations included in most commercial genetic testing profiles—while important for cystic fibrosis—may exclude key mutations related to pancreatitis. Finally, there are likely multiple CFTR associated mechanisms associated with pancreatitis and focusing on one or another alone may be misleading.
Over 1200 CFTR gene polymorphisms have been reported, and therefore identifying the important variants is challenging. These polymorphisms have been divided into five or six classes based on the functional consequences of the polymorphism on channel function.43–45 Class I–III mutations are severe (CFTRsev) and result in functional loss of CFTR from the epithelial cell surface. This is due to defects in biosynthesis, protein maturation, or channel regulation/gating. Class IV mutations are mild-variable mutations (CFTRm-v) and result in reduced, but not absence of, channel ion conductance. Currently, class V polymorphisms include mutations that diminish protein abundance or stability (also listed as class VI45,46) while class VI polymorphisms do not affect chloride conductance but rather the ability of CFTR to regulate other channels. Of special interest in pancreatic disease are a variety of variants thought to be associated with aberrant splicing or function of exons 9 and 10, including the intron 8 (IVS8) polyT tract (that is, the “5T” allele) and the IVS8 GT and TG microsatellites. The difficulty in evaluating these common variants is that they must be viewed within the context of all of the other mutations and variations with which they are commonly linked. This requires evaluation of the entire CFTR gene in large numbers of subjects and is rarely done in studies of pancreatitis.
The relationship between CFTR dysfunction and pancreatic disease appears to parallel the capacity of the pancreas to flush pancreatic digestive enzymes from the pancreatic duct. This is a multifactorial process in which CFTR plays a central but not exclusive role. As noted above, pancreatitis occurs following unregulated trypsinogen activation, zymogen activation, autodigestion, and subsequent inflammation. Activation of trypsinogen within the duct is especially dangerous as the trypsin autolysis mechanism is eliminated by elevated calcium. The most severe genetic risk is associated with the CFTRsev/CFTRsev genotype, which causes typical cystic fibrosis. Histologically, cystic fibrosis (of the pancreas) appears as typical chronic pancreatitis except for scattered protein filled ducts. Interestingly, signs of pancreatic involvement in cystic fibrosis47 correspond roughly to the gestational age of pancreatic trypsinogen expression, and elevated blood trypsinogen levels are used as a sign of pancreatic involvement of cystic fibrosis in screening programmes. Thus chronic pancreatitis of cystic fibrosis appears to develop in the context of trypsinogen expression and with an inability to clear digestive enzymes from the pancreatic duct.
Pancreatitis also occurs with the CFTRsev/CFTRm-v genotypes.33,34,41 These patients have “atypical cystic fibrosis” because the residual CFTR function, <10% of normal, provides enough anion transport for unstressed organ function.48 These patients are at increased risk of pancreatitis (for example, ∼80×49) but most patients with this genotype do not develop pancreatitis. As with SPINK1 mutations, recurrent acute pancreatitis and chronic pancreatitis appears to occur in the context of another environmental or genetic risk hit.
As noted above, recurrent acute pancreatitis and chronic pancreatitis also occurs in the context of heterozygous CFTR mutations. Three possible mechanisms are currently being explored to understand this observation. It could be that pancreatitis is associated with mutations that are not included in cystic fibrosis screening panels, and therefore these pancreatitis patients actually have a CFTRm-v/CFTRm-v genotype and atypical cystic fibrosis. Of note, there appears to be some common mutations in the CFTR molecule that preferentially alter bicarbonate conductance50–52 and would theoretically increase the risk of pancreatic injury compared with the risk of injury to other organs that rely on the chloride secreting properties of CFTR. These specific mutations appear to disrupt the function of exon 9 and 10 of the CFTR gene product. Another possibility is that CFTR is part of a complex disorder such as polygenic CFTR-SPINK1 pancreatitis in which heterozygous CFTR and heterozygous SPINK1 mutations cause pancreatitis when occurring together. Finally, some specific CFTR variants may increase susceptibility to environmental factors such as alcohol. Taken together, it appears that there are at least four possible types of CFTR associated pancreatitis. The first type is cystic fibrosis, the second atypical cystic fibrosis, and the third could be polygenic CFTR/geneB (for example, SPINK1) that increases the risk of recurrent acute pancreatitis. The fourth type is CFTR heterozygous mutations with strong metabolic or environmental factors such as excessive and prolonged alcohol intake. These latter two types remain theoretical. In addition, other factors may be important in enhancing or diminishing progressing to chronic inflammation and fibrosis.
Genetic testing for CFTR and SPINK1 mutations
Accurate, comprehensive, and predictive genetic testing for pancreatitis associated mutations in the SPINK1 gene and CFTR gene may be of great value in the future. The current limiting factor however is the lack of understanding of the mechanisms linking apparent heterozygous SPINK1 or CFTR mutations to pancreatitis. Without knowledge of the correct context, or other associated risk factors that are interacting with mutant CFTR or SPINK1, the value of a genetic test result is severely limited. Identification of heterozygous SPINK1 or CFTR mutations neither explains the disease in a person that has been diagnosed with pancreatitis nor accurately predicts the likelihood of pancreatitis in an unaffected person with concerns (for example, because of an affected relative or typical pain). Thus genetic testing for mutations in these two gene is currently considered premature.14–16
GENES ASSOCIATED WITH CHRONIC PANCREATITIS
If acute pancreatitis is viewed as an event, and chronic pancreatitis is an ongoing inflammatory process, then genetic and environmental factors influencing these to different degrees would also likely differ. Genetic, environmental, and structural factors that activate trypsinogen, prevent trypsin inhibition, or limit rapid clearance of active digestive enzymes form the pancreas increase susceptibility to acute and recurrent acute pancreatitis. In our prototype disease, hereditary pancreatitis, most individuals recover from acute pancreatitis through the normal healing process. However, a subset of people fail to recover and progress to chronic pancreatitis. From our perspective, determinants of the risk of progressing from recurrent acute pancreatitis to chronic pancreatitis are different from those that determine susceptibility to acute pancreatitis, and include environmental, immunological, and other factors. These factors have been organised into the SAPE hypothesis model (fig 3 ▶) and is discussed in detail elsewhere.53 Several interesting genetic polymorphisms have been described that may contribute to chronic injury or fibrosis. Alcohol may be especially important in pancreatic disease because it is a risk factor for several hypothesised steps in the same overall process.
Figure 3.
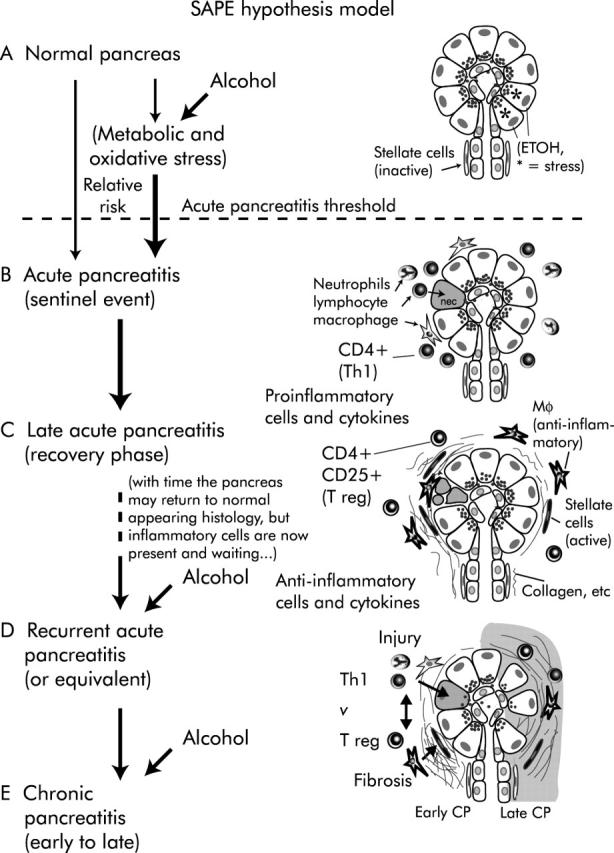
SAPE (sentinel acute pancreatitis event) hypothesis model. The sequence of events leading to chronic pancreatitis in the SAPE hypothesis model. (A) Normal pancreas. If the subject is a heavy alcohol user, acinar cells are under metabolic and oxidative stress (*) but histology remains relatively normal. Alcohol increases the risk of crossing the acute pancreatitis threshold (bold arrow crossing the broken line). (B) Acute pancreatitis with pancreatic injury and infiltration of proinflammatory cells. The first or “sentinel” acute pancreatitis event (SAPE) is a critical step because it initiates the inflammatory process that results in both injury and later fibrosis. (C) Late acute pancreatitis is dominated by anti-inflammatory cells that limit further injury by proinflammatory cells and products, and promote healing. This includes activation of stellate cells with produce collagen, etc. In the absence of recurrent acute pancreatitis, acinar cell toxins (for example, high dose alcohol), or factors that activate the immune system, the pancreas may eventually return to normal appearing histology except for some residual inflammatory cells that are primed to respond to any future injury. (D) Recurrent acute pancreatitis, acinar cell injury, or other factors that activate an acute inflammatory response (Th1) are immediately countered by an anti-inflammatory counter response (T reg) which, among other things, drives fibrosis. This vicious cycle results in both continued injury (top) and further fibrosis (bottom) leading to (E) extensive acinar cell loss and sclerosis (right) that is characteristic of chronic pancreatitis. Both genetic factors and environmental factors play a role in this process by increasing susceptibility to acute pancreatitis, altering the severity and duration of acute pancreatitis, and altering the healing processes that drive fibrosis. Alcohol is especially important because it acts at multiple steps in this process.
UDP glucuronosyltransferase (UGT1A7)
Ockenga and colleagues54 recently reported an increased risk for chronic alcoholic pancreatitis in patients with mutations in uridine 5′-diphosphate glucuronosyltransferase (UGT). UDP glucuronosyltransferases are a family of phase II enzymes involved in metabolism, detoxification, and other aspects of cellular defence.54 Of the 10 major isoforms in this superfamily, only the UGT1A7 gene is expressed in high levels in the pancreas along with small amounts of UGT1A4 and trace amounts of UGT1A3.54 The UGT1A7 gene is known to have five common single nucleotide polymorphisms which are linked as three polymorphic UGT1A7 alleles (UGT1A7*2, UGT1A7*3, and UGT1A7*4).54 The UGT1A7*3 haplotype results in a significant decrease in UGT1A7 enzyme activity. The UGT1A7*3 allele is common, appearing in 21% of the control population but was found in 37% of alcoholic pancreatitis patients (89% smokers), which was significantly more common (p = 0.001) and more than doubled the risk of chronic pancreatitis (odds ratio 2.24). Interestingly, there was also a trend toward chronic pancreatitis in non-alcoholic subjects with the UGT1A7*3 allele that were SPINK1 N34S negative (p = 0.08; odds ratio 1.82), but not in patients the were SPINK1 N34S positive (p = 0.27; 1.33). This last finding may reflect the early median age of disease onset of SPINK1 N34S patients versus non-alcoholic SPINK mutation negative patients (19 (14) years v 33 (11) years—see Pfützer and colleagues32) who were also less likely to be tobacco smokers (6% v 42%). This finding represents one of the first examples of a common polymorphism in a drug metabolising gene associated with chronic pancreatitis. It was not clear from this study however if the gene was associated with alcohol or tobacco smoking as most alcoholic patients smoked. These findings have not yet been confirmed in other populations, and the risk is relatively low (odds ratio 2.24), and hence genetic testing for this allele is currently not recommended.
Alcohol metabolising genes
Mutations in the major alcohol metabolising genes do not appear to be associated with (alcoholic) chronic pancreatitis.55–57 This includes alcohol dehydrogenase 2 and aldehyde dehydrogenase 2 in Caucasians—with conflicting reports in Japanese— alcohol dehydrogenase 3, cytochrome P450-2E1 (CYP2E1), glutathione S-transferase-M1 (GSTM1), GSTT1, etc.57–59 Chronic alcohol consumption appears to lower the threshold for initiating acute pancreatitis60 and alcohol is a major cause of acute pancreatitis in adults, but the relationship between alcohol consumption and chronic pancreatitis is complex and multifactorial.55
Investigators continue to revisit the role of the GST family of genes (for example, MGST1, GSTM3, GSTT1, and GSTM1) for pancreatitis associated mutations, looking specifically at subsets of pancreatic disease.61,62 Although no association has been identified between genetic polymorphisms and hereditary, idiopathic or alcoholic pancreatitis in general,61,63 the GSTM1 null genotypes (no gene product expressed) appeared to protect a small subset of individuals from alcoholic chronic pancreatitis—that is, women under the age of 50 years.62 The reason for this last observation is unknown but could reflect more severe pancreatitis in subjects who are not alcoholic (and therefore have normal glutathione stores) and who have an exaggerated immune response and injury associated with normal GSTM1 genotypes. However, no clear mechanism has yet been described.
Immune and fibrosis modulating genes
Cavestro et al recently reported an association between the HLA-DRB1*0401 allele and chronic pancreatitis.64 The DRB1*04 allele was significantly associated with chronic pancreatitis (26.78% v 8.1%; p<0.003; odds ratio 4.1; confidence interval 1.85−9.06), and specifically the DRB1*0401 allele (14.3% v 1.1%; p = 0.00017; odds ratio 15.08; confidence interval 3.1−73.36). Cavestro et al also reported an association between mutations in the keratin 8 gene and chronic pancreatitis.65 A keratin 8 gene G61C mutation was identified in six of 67 patients with chronic pancreatitis but not in 100 controls (8.9 v 0%; p<0.003; odds ratio 21.24; confidence interval 2.74−164.42). The importance of this finding is yet to be determined because it has not yet been confirmed by others, and we do not see this association in our American populations (unpublished).
A number of cytokines, including tumour necrosis factor α (TNF-α) and the interleukins 1, 6, and 10 (IL-1, IL-6, and IL-10) appear to be important in modulating fibrosis of chronic pancreatitis through actions on the pancreatic stellate cells.66 Mutations in the promoter or coding region of TNF-α and the TNF receptor gene do not appear to act as susceptibility genes for hereditary, familial, idiopathic chronic pancreatitis or alcoholic chronic pancreatitis,67,68 but the TNF-238A promoter polymorphism may be modifier gene accelerating chronic pancreatitis in patients with underlying trypsinogen mutations.68 Many of the IL-10 gene polymorphisms have already been excluded as playing the dominant factor leading to chronic pancreatitis68,69 while other cytokines continue to be investigated.
Risk factors and environmental factors important to chronic pancreatitis can be viewed together (fig 4 ▶). In this paradigm, the factor that is triggering and driving chronic pancreatitis is recurrent acute pancreatitis. If the number of episodes of acute pancreatitis was plotted (fig 4 ▶, x axis) and compared with fibrosis, current evidence would predict a proportional degree of fibrosis (fig 4 ▶, y axis). Variation from this predicted relationship would reflect the influence of other modifying risk factors (more fibrosis than predicted by the number of acute pancreatitis attacks) or protective factors (less fibrosis than predicted by the number of acute pancreatitis attacks). Although such a relationship has not yet been demonstrated in pancreatitis, it offers a conceptual framework for future investigation and testing.
Figure 4.

Modifier genes and factors effecting chronic pancreatitis. Hypothetical model illustrating the relationship between recurrent acute pancreatitis (RAP) or other immune system activating events (x axis) and the development of pancreatic fibrosis (y axis) which serves as a marker of severity of chronic pancreatitis. According to the SAPE hypothesis model, recurrent episodes of acute pancreatitis will lead to pancreatic fibrosis (average risk—solid line). Subjects with polymorphisms in modifier genes or who have other risk factors (chronic pancreatitis (CP) modifier genes—upper broken line) develop more fibrosis with less RAP, while patients with protective genes and factors (lower broken line) develop minimal fibrosis despite frequent RAP.
ISSUES IN GENETIC TESTING
The purpose of genetic testing can be divided into two general categories: diagnostic and predictive. Diagnostic testing is done when a patient has symptoms of a disease and a genetic test is done to determine the underlying cause(s). Examples include CFTR mutation testing in suspected typical or atypical cystic fibrosis, or PRSS1 (cationic trypsinogen) gene testing in suspected hereditary pancreatitis. Predictive testing is genetic testing in subjects without evidence of pancreatic disease. For example, an affected parent within a PRSS1 R122H hereditary pancreatitis kindred may want to know if their child carries the R122H mutation, or an unaffected sibling of an affected individual may want to know if they are a carrier for reproductive or lifestyle decisions. In both cases the physician should understand the implications of testing based on proper interpretation of positive or negative results. Before a genetic test of any type is considered, one must anticipate the potential magnitude of the results on the patient and their family and determine whether formal genetic counselling is necessary. The need for formal genetic counselling however depends on the complexity of the personal implications of the test result, and some simple genetic risk factors will likely become a routine part of clinical practice without formal pretest counselling.1 Testing for polygenic or complex genetic disorders will likely fall into the latter category as the implications for family and other social issues is minimised.
CONCLUSIONS
Since the discovery that mutations in the cationic trypsinogen gene caused hereditary pancreatitis in 199617 it has become increasingly evident that acute and chronic pancreatitis are syndromes encompassing several complex disease mechanisms. The key genetic, environmental, and morphological factors that intersect to cause a complex pancreatic disease are slowly emerging, and knowledge of these factors will allow for disease models to be developed. These models will raise new research questions that alter current therapy and may limit progression of early disease.
Ongoing clarification of which polymorphisms confer susceptibility to acute pancreatitis, and which polymorphisms modify the immunological response to acute and recurrent acute pancreatitis that lead to chronic pancreatitis will also be important in determining the timing of genetic testing and subsequent intervention. Genetic testing of the future will therefore require inclusion of many polymorphisms, perhaps hundreds, and the results will require careful interpretation and educational information. It is conceivable that therapy will be targeted at preventing acute pancreatitis in some cases whereas in other cases therapy will be targeted at metabolic stress or the immune response. If chronic pancreatitis is indeed the result of multifactorial processes, and if the factors and pathological pathways were identified early, then modifiable risk should be identified and addressed early so that rational preventative measures can be taken.
Acknowledgments
The author is indebted to M Michael Barmada PhD and Scott Plevy MD for critical review of this manuscript, and to Andrew Brunskill PhD for figure 1 ▶. This work was supported by NIH grants DK54709 and DK061451 and the Cystic Fibrosis Foundation.
Conflict of interest: Dr Whitcomb owns the patent for trypsinogen gene testing in the United States which is licensed to Ambry Genetics.
REFERENCES
- 1.Burke W . Genetic testing. N Engl J Med 2002;347:1867–75. [DOI] [PubMed] [Google Scholar]
- 2.Applebaum SE, Kant JA, Whitcomb DC, et al. Genetic testing: counseling, laboratory and regulatory issues and the EUROPAC protocol for ethical research in multi-center studies of inherited pancreatic diseases. Med Clin North Am 2000;82:575–88. [DOI] [PubMed] [Google Scholar]
- 3.Applebaum SE, O’Connell JA, Aston CE, et al. Motivations and concerns of patients with access to genetic testing for hereditary pancreatitis. Am J Gastroenterol 2001;96:1610–17. [DOI] [PubMed] [Google Scholar]
- 4.Shaheen NJ, Lawrence LB, Bacon BR, et al. Insurance, employment, and psychosocial consequences of a diagnosis of hereditary hemochromatosis in subjects without end organ damage. Am J Gastroenterol 2003;98:1175–80. [DOI] [PubMed] [Google Scholar]
- 5.Whitcomb DC. Early trypsinogen activation in acute pancreatitis. Gastroenterology 1999;116:770–3. [PubMed] [Google Scholar]
- 6.Lerch MM, Gorelick FS. Early trypsinogen activation in acute pancreatitis. Med Clin North Am 2000;84:549–63. [DOI] [PubMed] [Google Scholar]
- 7.Raraty M , Ward J, Erdemli G, et al. Calcium-dependent enzyme activation and vacuole formation in the apical granular region of pancreatic acinar cells. Proc Natl Acad Sci U S A 2000;97:13126–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruger B , Albrecht E, Lerch MM. The role of intracellular calcium signaling in premature protease activation and the onset of pancreatitis. Am J Pathol 2000;157:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voronina S , Longbottom R, Sutton R, et al. Bile acids induce calcium signals in mouse pancreatic acinar cells: implications for bile-induced pancreatic pathology. J Physiol 2002;540:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon P , Weiss FU, Sahin-Tóth M, et al. Hereditary pancreatitis caused by a novel PRSS1 mutation (Arg-122->Cys) that alters autoactivation and autodegradation of cationic trypsinogen. J Biol Chem 2001;21:21. [DOI] [PubMed] [Google Scholar]
- 11.Colomb E , Guy O, Deprez P, et al. The two human trypsinoens: catalytic properties of the corresponding trypsins. Biochem Biophys Acta 1978;525:186–93. [DOI] [PubMed] [Google Scholar]
- 12.Frick TW, Fernandez-del CC, et al.Elevated calcium and activation of trypsinogen in rat pancreatic acini. Gut 1997;41:339–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutton R , Criddle D, Raraty MG, et al. Signal transduction, calcium and acute pancreatitis. Pancreatology 2003;3:497–505. [DOI] [PubMed] [Google Scholar]
- 14.Ellis I , Lerch MM, Whitcomb DC, et al. Genetic testing for hereditary pancreatitis: Guidelines for indications, counseling, consent and privacy issues. Pancreatology 2001;1:401–11. [DOI] [PubMed] [Google Scholar]
- 15.Whitcomb DC. Motion-genetic testing is useful in the diagnosis of nonhereditary pancreatic conditions: Arguments for the motion. Can J Gastroenterol 2003;17:47–52. [DOI] [PubMed] [Google Scholar]
- 16.Cohn JA. Motion-genetic testing is useful in the diagnosis of nonhereditary pancreatic conditions: Arguments against the motion. Can J Gastroenterol 2003;17:53–5. [DOI] [PubMed] [Google Scholar]
- 17.Whitcomb DC, Gorry MC, Preston RA, et al. Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996;14:141–5. [DOI] [PubMed] [Google Scholar]
- 18.Applebaum-Shapiro SE, Finch R, Pfützer RH, et al. Hereditary pancreatitis in North America: The Pittsburgh-Midwest Multi-Center Pancreatic Study Group Study. Pancreatology 2001;1:439–43. [DOI] [PubMed] [Google Scholar]
- 19.Whitcomb DC. Hereditary pancreatitis: New insights into acute and chronic pancreatitis. Gut 1999;45:317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahin-Tóth M , Tóth M. Gain-of-function mutations associated with hereditary pancreatitis enhance autoactivation of human cationic trypsinogen. Biochem Biophys Res Commun 2000;278:286–9. [DOI] [PubMed] [Google Scholar]
- 21.Sibert JR. Hereditary pancreatitis in England and Wales. J Med Genet 1978;15:189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Bodic L , Schnee M, Georgelin T, et al. An exceptional genealogy for hereditary chronic pancreatitis. Dig Dis Sci 1996;41:1504–10. [DOI] [PubMed] [Google Scholar]
- 23.Sossenheimer MJ, Aston CE, Preston RA, et al. Clinical characteristics of hereditary pancreatitis in a large family based on high-risk haplotype. Am J Gastroenterol 1997;92:1113–6. [PubMed] [Google Scholar]
- 24.Lowenfels A , Maisonneuve P, DiMagno E, et al. Hereditary pancreatitis and the risk of pancreatic cancer. J Natl Cancer Inst 1997;89:442–6. [DOI] [PubMed] [Google Scholar]
- 25.Howes N , Wong T, Greenhalf W, et al. Pancreatic cancer risk in hereditary pancreatitis in Europe. Digestion 2000;61:300. [Google Scholar]
- 26.Amann ST, Gates LK, Aston CE, et al. Expression and penetrance of the hereditary pancreatitis phenotype in monozygotic twins. Gut 2001;48:542–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Teich N , Mössner J, Keim V. Systematac overview of genetic variants of cationic trypsinogen and SPINK1 in pancreatitis patients. In: Durie P, Lerch MM, Lowenfels AB, et al, eds. Genetic disorders of the exocrine pancreas: an overview and update. Basel: Karger, 2002:20–2.
- 28.Whitcomb DC. Hereditary diseases of the pancreas. In: Yamada T, Albers DH, Laine L, et al, eds. Textbook of gastroenterology. Philadelphia: Lippincott, Williams and Wilkins, 2003:2147–65.
- 29.Comfort M , Steinberg A. Pedigree of a family with hereditary chronic relapsing pancreatitis. Gastroenterology 1952;21:54–63. [PubMed] [Google Scholar]
- 30.Lowenfels AB, Maisonneuve P, Whitcomb DC, et al. Cigarette smoking as a risk factor for pancreatic cancer in patients with hereditary pancreatitis. J Am Med Assoc 2001;286:169–70. [DOI] [PubMed] [Google Scholar]
- 31.Witt H , Luck W, Hennies HC, et al. Mutations in the gene encoding the serine protease inhibitor, kazal type 1 are associated with chronic pancreatitis. Nat Genet 2000;25:213–16. [DOI] [PubMed] [Google Scholar]
- 32.Pfützer RH, Barmada MM, Brunskil APJ, et al. SPINK1/PSTI polymorphisms act as disease modifiers in familial and idiopathic chronic pancreatitis. Gastroenterology 2000;119:615–23. [DOI] [PubMed] [Google Scholar]
- 33.Sharer N , Schwarz M, Malone G, et al. Mutations of the cystic fibrosis gene in patients with chronic pancreatitis. N Engl J Med 1998;339:645–52. [DOI] [PubMed] [Google Scholar]
- 34.Cohn JA, Friedman KJ, Noone PG, et al. Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 1998;339:653–8. [DOI] [PubMed] [Google Scholar]
- 35.Whitcomb DC. How to think about SPINK and pancreatitis. Am J Gastroenterol 2002;97:1085–8. [DOI] [PubMed] [Google Scholar]
- 36.Threadgold J , Greenhalf W, Ellis I, et al. The N34S mutation of SPINK1 (PSTI) is associated with a familial pattern of idiopathic chronic pancreatitis but does not cause the disease. Gut 2002;50:675–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen J-M, Mercier B, Audrezet M-P, et al. Mutational analysis of the human pancreatic secretory trypsin inhibitor (PSTI) gene in hereditary and sporadic chronic pancreatitis. J Med Genet 2000;37:67–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfutzer RH, Whitcomb DC. SPINK1 mutations are associated with multiple phenotypes. Pancreatology 2001;1:457–60. [DOI] [PubMed] [Google Scholar]
- 39.Hassan Z , Mohan V, Ali L, et al. SPINK1 is a susceptibility gene for fibrocalculous pancreatic diabetes in subjects from the Indian subcontinent. Am J Hum Genet 2002;71:964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felderbauer P , Hoffmann P, Einwachter H, et al. A novel mutation of the calcium sensing receptor gene is associated with chronic pancreatitis in a family with heterozygous SPINK1 mutations. BMC Gastroenterol 2003;3:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noone PG, Zhou Z, Silverman LM, et al. Cystic fibrosis gene mutations and pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene mutations. Gastroenterology 2001;121:1310–19. [DOI] [PubMed] [Google Scholar]
- 42.Norton ID, Apte MV, Dixson H, et al. Cystic fibrosis genotypes and alcoholic pancreatitis. J Gastroenterol Hepatol 1998;13:496–9. [DOI] [PubMed] [Google Scholar]
- 43.Mickle JE, Cutting GR. Genotype-phenotype relationships in cystic fibrosis. Med Clin North Am 2000;84:597–607. [DOI] [PubMed] [Google Scholar]
- 44.Zielenski J , Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet 1995;29:777–807. [DOI] [PubMed] [Google Scholar]
- 45.Rowntree RK, Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet 2003;67:471–85. [DOI] [PubMed] [Google Scholar]
- 46.Zielenski J . Genotype and phenotype in cystic fibrosis. Respiration 2000;67:117–33. [DOI] [PubMed] [Google Scholar]
- 47.Imrie JR, Fagan DG, Sturgess JM. Quantitative evaluation of the development of the exocrine pancreas in cystic fibrosis and control infants. Am J Pathol 1979;95:697–707. [PMC free article] [PubMed] [Google Scholar]
- 48.Stern RC. The diagnosis of cystic fibrosis. N Engl J Med 1997;336:487–91. [DOI] [PubMed] [Google Scholar]
- 49.Cohn JA, Bornstein JD, Jowell PS. Cystic fibrosis mutations and genetic predisposition to idiopathic chronic pancreatitis. Med Clin North Am 2000;84:621–31. [DOI] [PubMed] [Google Scholar]
- 50.Ko SB, Shcheynikov N, Choi JY, et al. A molecular mechanism for aberrant CFTR-dependent HCO(3)(−) transport in cystic fibrosis. EMBO J 2002;21:5662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi JY, Muallem D, Kiselyov K, et al. Aberrant CFTR-dependent HCO3− transport in mutations associated with cystic fibrosis. Nature 2001;410:94–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reddy MM, Quinton PM. Control of dynamic CFTR selectivity by glutamate and ATP in epithelial cells. Nature 2003;423:756–60. [DOI] [PubMed] [Google Scholar]
- 53.Schneider A , Whitcomb DC. Hereditary pancreatitis: a model for inflammatory diseases of the pancreas. Best Pract Res Clin Gastroenterol 2002;16:347–63. [DOI] [PubMed] [Google Scholar]
- 54.Ockenga J , Vogel A, Teich N, et al. UDP Glucuronosyltransferase (UGT1A7) gene polymorphisms increase the risk of chronic pancreatitis and pancreatic cancer. Gastroenterology 2003;124:1802–8. [DOI] [PubMed] [Google Scholar]
- 55.Etemad B , Whitcomb DC. Chronic pancreatitis: Diagnosis, classification, and new genetic developments. Gastroenterology 2001;120:682–707. [DOI] [PubMed] [Google Scholar]
- 56.Hanck C , Schneider A, Whitcomb DC. Genetic polymorphisms in alcoholic pancreatitis. Best Pract Res Clin Gastroenterol 2003;17:613–23. [DOI] [PubMed] [Google Scholar]
- 57.Frenzer A , Butler WJ, Norton ID, et al. Polymorphism in alcohol-metabolizing enzymes, glutathione S-transferases and apolipoprotein E and susceptibility to alcohol-induced cirrhosis and chronic pancreatitis. J Gastroenterol Hepatol 2002;17:177–82. [DOI] [PubMed] [Google Scholar]
- 58.Maruyama K , Takahashi H, Matsushita S, et al. Genotypes of alcohol-metabolizing enzymes in relation to alcoholic chronic pancreatitis in Japan. Alcohol Clin Exp Res 1999;23 (suppl 4) :85–91S. [DOI] [PubMed] [Google Scholar]
- 59.Yang B , O’Reilly DA, Demaine AG, et al. Study of polymorphisms in the CYP2E1 gene in patients with alcoholic pancreatitis. Alcohol 2001;23:91–7. [DOI] [PubMed] [Google Scholar]
- 60.Pandol SJ, Periskic S, Gukovsky I, et al. Ethanol diet increases the sensitivity of rats to pancreatitis induced by cholecystokinin octapeptide. Gastroenterology 1999;117:706–16. [DOI] [PubMed] [Google Scholar]
- 61.Bartsch H , Malaveille C, Lowenfels AB, et al. Genetic polymorphism of N-acetyltransferases, glutathione S-transferase M1 and NAD(P)H:quinone oxidoreductase in relation to malignant and benign pancreatic disease risk. The International Pancreatic Disease Study Group. Eur J Cancer Prev 1998;7:215–23. [DOI] [PubMed] [Google Scholar]
- 62.Verlaan M , Te Morsche RH, Roelofs HM, et al. Glutathione S-transferase mu null genotype affords protection against alcohol induced chronic pancreatitis. Am J Med Genet 2003;1:34–9. [DOI] [PubMed] [Google Scholar]
- 63.Schneider A , Tögel S, Barmada MM, et al. Genetic analysis of the glutathione s-transferase genes MGST1, GSTM3, GSTT1 and GSTM1 in patients with hereditary pancreatitis. J Gastroenterol 2004; (in press). [DOI] [PubMed]
- 64.Cavestro GM, Frulloni L, Neri TM, et al. Association of HLA-DRB1*0401 allele with chronic pancreatitis. Pancreas 2003;26:388–91. [DOI] [PubMed] [Google Scholar]
- 65.Cavestro GM, Frulloni L, Nouvenne A, et al. Association of keratin 8 gene mutation with chronic pancreatitis. Dig Liver Dis 2003;35:416–20. [DOI] [PubMed] [Google Scholar]
- 66.Mews P , Phillips P, Fahmy R, et al. Pancreatic stellate cells respond to inflammatory cytokines: potential role in chronic pancreatitis. Gut 2002;50:535–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schneider A , Pogue-Geile K, Barmada MM, et al. Hereditary, familial, and idiopathic chronic pancreatitis are not associated with polymorphisms in the tumor necrosis factor alpha (TNF-alpha) promoter region or the TNF receptor 1 (TNFR1) gene. Genet Med 2003;5:120–5. [DOI] [PubMed] [Google Scholar]
- 68.Beranek H , Teich N, Witt H, et al. Analysis of tumour necrosis factor alpha and interleukin 10 promotor variants in patients with chronic pancreatitis. Eur J Gastroenterol Hepatol 2003;15:1223–7. [DOI] [PubMed] [Google Scholar]
- 69.Schneider A , Barmada MM, Slivka A, et al. Analysis of tumor necrosis factor-alpha, transforming growth factor-beta1, interleukin-10, and interferon-gamma polymorphisms in patients with alcoholic chronic pancreatitis. Alcohol 2004;32:19–24. [DOI] [PubMed] [Google Scholar]