

Abstract
Background: Folylpoly-γ-glutamate synthetase (FPGS) converts intracellular folates and antifolates (for example, methotrexate (MTX)) to polyglutamates. Polyglutamylated folates and antifolates are retained in cells longer and are better substrates than their monoglutamate counterparts for enzymes involved in one carbon transfer. Polyglutamylation of intracellular 5,10-methylenetetrahydrofolate may also enhance the cytotoxicity of 5-fluorouracil (5-FU) by allowing more efficient formation and stabilisation of the inhibitory ternary complex involving thymidylate synthase and a 5-FU metabolite.
Aim: We investigated the effects of FPGS modulation on the chemosensitivity of colon cancer cells to 5-FU and MTX.
Methods: Human HCT116 colon cancer cells were stably transfected with the sense or antisense FPGS cDNA or blank (control). FPGS protein expression and enzyme activity, growth rate, intracellular folate content and composition, and in vitro chemosensitivity to 5-FU and MTX were determined.
Results: Compared with cells expressing endogenous FPGS, those overexpressing FPGS had significantly faster growth rates and higher concentrations of total folate and long chain folate polyglutamates while antisense FPGS inhibition produced opposite results. FPGS overexpression significantly enhanced, whereas FPGS inhibition decreased, chemosensitivity to 5-FU. No significant difference in chemosensitivity to MTX was observed.
Conclusions: These data provide functional evidence that FPGS overexpression and inhibition modulate chemosensitivity of colon cancer cells to 5-FU by altering intracellular folate polyglutamylation, providing proof of principle. Thus FPGS status may be an important predictor of chemosensitivity of colon cancer cells to 5-FU based chemotherapy, and FPGS gene transfer may increase the sensitivity of colon cancer cells to 5-FU-based chemotherapy.
Keywords: folylpolyglutamate synthetase, colon cancer, chemotherapy, 5-fluorouracil, methotrexate
Folate mediates the transfer of one carbon units necessary for the de novo biosynthesis of purines and thymidylate, and hence is an essential factor for DNA synthesis.1 While monoglutamates are the only circulating forms of folate in blood and the only form of folate that is transported across the cell membrane, once taken up into cells, intracellular folate exists primarily as polyglutamates.1 Intracellular folate is converted to polyglutamates by folylpolyglutamate synthetase (FPGS), while γ-glutamyl hydrolase (GGH) removes the terminal glutamates.1 Polyglutamylated folates are better retained in cells and are better substrates than monoglutamates for intracellular folate dependent enzymes.2
Similarly, antifolates (for example, methotrexate (MTX)) are retained in tumour and normal cells by FPGS induced polyglutamylation, and are exported from cells after hydrolysis to monoglutamates by GGH.2,3 As with folate, polyglutamylated antifolates are retained in cells longer, thereby increasing their cytotoxicity by extending the length of exposure.4–6 Polyglutamylated antifolates generally have a higher affinity for and hence inhibit their target folate dependent enzymes in the thymidylate and purine biosynthetic pathways (for example, thymidylate synthase (TS)) to a greater extent than the monoglutamate forms.7,8,9,10,11 Decreased antifolate polyglutamylation due to quantitative12–15 or qualitative16–18 alterations in FPGS activity has been shown to be a mechanism of resistance to MTX and other antifolates in human and murine leukaemia cell lines. Transfection of FPGS cDNA has been shown to increase sensitivity to MTX in variant hamster cells that lack endogenous FPGS activity.19 FPGS gene transfer into rat and human glioma, gliosarcoma, and glioblastoma cell lines already expressing FPGS significantly enhanced sensitivity to MTX and other antifolates.20
FPGS induced polyglutamylation may also affect the sensitivity of tumour cells to other chemotherapeutic agents not typically considered as antifolates such as 5-fluorouracil (5-FU). One cytotoxic mechanism of 5-FU is the formation of a ternary complex involving 5-fluoro-2-deoxyuridine-5-monophosphate (FdUMP; a metabolite of 5-FU), TS, and 5,10-methylenetetrahydrofolate (5,10-methyleneTHF), thereby inhibiting TS activity with consequent depletion of intracellular thymidylate and ultimately suppression of DNA synthesis.21 Leucovorin (LV), a precursor for 5,10-methyleneTHF, potentiates the cytotoxic effect of 5-FU by stabilising the inhibitory 5,10-methyleneTHF-TS-FdUMP ternary complex.21 5,10-MethyleneTHF with longer chain length polyglutamates is better retained intracellularly and is more efficient in the formation and stabilisation of the inhibitory 5,10-methyleneTHF-TS-FdUMP ternary complex compared with shorter chain polyglutamates.22 Decreased FPGS activity has previously been demonstrated to confer resistance to 5-FU in some human cancer cell lines.23–25
FPGS appears to play an important role in the sensitivity of cancer cells to antifolates and 5-FU and thus FPGS modulation might be a potential therapeutic target for increasing sensitivity of cancer cells to these chemotherapeutic agents. However, definitive functional evidence supporting the effect of FPGS modulation on the chemosenstivity of colon cancer cells to MTX and 5-FU is currently lacking. Therefore, we generated an in vitro model of FPGS modulation in colon cancer cells and determined the effect of FPGS overexpression and inhibition on the chemosensitivity of colon cancer cells to MTX and 5-FU.
MATERIALS AND METHODS
Cell line and culture
Human colon adenocarcinoma HCT116 cells were purchased from the American Type Culture Collection (Manassas, Virginia, USA). HCT116 cells exhibit microsatellite instability due to an inactivating MLH1 mutation, contain a k-ras protocongene mutation, and lack p53 and APC mutations. Cells were grown in RPMI-1640 medium (Invitrogen, Gaithersburg, Maryland, USA) supplemented with 10% fetal bovine serum, 2 mM l-glutamine, penicillin 100 U/ml, and streptomycin 100 mg/ml. Cultures were maintained at 37°C in 5% CO2.
Construction and transfection of the sense and antisense FPGS expression vector
The full length human FPGS cDNA was provided by Dr A Bognar (University of Toronto, Toronto, Canada). The full length human FPGS cDNA was subcloned into the EcoRI site of the eukaryotic expression vector pIRESneo (Clontech, Palo Alto, California, USA) containing a CMV promoter and a neomycin resistance gene expression cassette in the sense and antisense orientation to generate the sense and antisense FPGS expression vectors, respectively. Correct integration, orientation, and sequence of the sense and antisense FPGS cDNAs were confirmed by predicted fragment sizes after multiple restriction enzyme digestions and DNA sequencing. The pIRESneo vector containing the sense or antisense FPGS cDNA was stably transfected into HCT116 cells using Lipofectin (Invitrogen) according to the manufacturer’s protocol. In a separate transfection, HCT116 cells were stably transfected with empty pIRESneo vector (endogenous FPGS). Transfected cells were incubated with 500 μg/ml of neomycin (Invitrogen) to select for cells that expressed the various constructs. After a population of cells was selected, individual clonal cell lines were isolated and expanded. Cells were maintained in complete medium supplemented with neomycin 500 μg/ml. Several (>10) clones expressing the sense and antisense FPGS cDNA and empty vector were screened at random, and two independent clones of each construct were selected for further analyses. Data from two experiments using two independent clones of each construct were similar and thus the data from one experiment are presented.
Western blot analysis
FPGS, TS, p53, and p21 protein expression was determined by standard western analysis, as described previously,26 using a rabbit polyclonal antibody raised against a peptide sequence spanning amino acids 275–290 of human FPGS27 (Zymed, San Francisco, California, USA) at a dilution of 1:200, a sheep polyclonal antibody against human TS (Rockland Immunochemicals, Gilbertsville, Pennsylvania, USA) at a dilution of 1:3000, and anti-p53(DO-1) and p21 monoclonal antibody (Oncogene Research Products, Boston, Massachusetts, USA) at a dilution of 1:3000, respectively. All western analyses were repeated using three different cell lysates.
FPGS and TS activity assay
FPGS activity was determined by measuring incorporation of [3H]glutamate into the polyglutamate chain of aminopterin, as described previously.20 The catalytic activity of TS was determined by 3H release that occurred during conversion of [5-3H]-deoxyuridine-5-monophosphate to deoxythymidine-5-monophosphate, as described previously.28 Enzyme assays were performed in triplicate and repeated using three different cell lysates.
Intracellular folate concentrations and determination of glutamate chain lengths
Intracellular folate concentrations were determined by a standard microbiological microtitre plate assay using Lactobacillus casei29 for both conjugase treated and untreated samples to determine the extent of polyglutamylation as L casei grow in proportion to the amount of mono-, di-, tri- and, to some extent, tetra-glutamylated folate in the samples, and conjugase treatment results in cleavage of the polyglutamylated chain to yield these short chain length forms of folate. This method has been used in previous studies to determine overall pool of long chain versus short chain polyglutamates.30 All analyses were performed in triplicate and repeated using three different cell lysates.
Real time quantitative reverse transcription-PCR
Total cellular RNA was extracted using the RNeasy MidiKit (Qiagen, Mississauga, Ontario, Canada). cDNA was generated from total RNA using random primers and the SuperScript III RNase H-Reverse Transcriptase (Invitrogen). Polymerase chain reaction (PCR) primers for GGH were constructed based on the human GGH cDNA sequence31 and were synthesised by ACGT (Toronto, Ontario, Canada) as follows: forward, 5′-GCC ACA GAT ACT GTT GAC GTG G-3′; reverse, 5′-ATG GAA ATT GGC AGT CAG AGG-3′. Real time PCR was performed using the LC FastStart DNA Master SYBR Green 1 Kit in the LightCycler rapid thermal cycler system (Roche Diagnostics, Laval, Quebec, Canada) and the melting curve was determined as described.32 All PCR reactions were performed in triplicate, and experiments were repeated three times.
Doubling time calculation
Cells (8000 per well) were plated in 96 well plates and grown in RPMI-1640 medium with 10% fetal bovine serum for 72 hours. The cell population was determined using the sulforhodamine B (SRB) optical density (OD) measurement assay. The growth rate constant k was derived using the equation
N/N0 = ekt
where N0 is the OD of cells at time zero and N is the OD of cells at 72 hours. The same equation was used to calculate the doubling time t by setting N/N0 = 2. All analyses were performed in triplicate, and three replicate experiments were performed.
In vitro chemosensitivity assay
In vitro chemosensitivitiy was determined using a modification of the SRB protein assay, as described previously.26,29,33 Briefly, 8000 cells per 100 μl RPMI-1640 medium per well were seeded in triplicate in 96 well flat bottom plates (Costar, Cambridge, Massachusetts, USA). After 24 hours, an additional 100 μl of RPMI-1640 medium containing MTX (Schircks, Jona, Switzerland) or 5-FU (InvivoGen, San Diego, California, USA) in combination with LV (Sigma Aldrich Canada, Oakville, Ontario, Canada) were added, and cells were cultured for an additional 72 hours. The concentration of 5-FU was varied, with concentrations ranging from 1.5×10−6 M to 25×10−6 M, whereas the concentration of LV was held constant at 5×10−6 M. LV was added to simulate the standard 5-FU based chemotherapy used in the treatment of colorectal cancer.21 The concentration of MTX was varied, with concentrations ranging from 3.5×10−9 M to 5×10−8 M. After 72 hours, cells were fixed with trichloroacetic acid and stained with SRB protein dye. The dye was solubilised, and the OD of the solution measured at 595 nm. The results were expressed as percentage of cell survival on the basis of the difference between the OD at the start and end of drug exposure, according to the formula:
Survival = [(ODdrug/ODstart drug exposure) − 1] / [(ODno drug/ODstart drug exposure) −1] ×100%.
IC25, IC50, and IC75 values (that is, the drug concentration that corresponded to a reduction in cell survival by 75%, 50%, and 25%, respectively, compared with survival of untreated control cells) were calculated from plots of drug concentration versus proportion of cells that survived. All analyses were performed in triplicate, and three replicate experiments were performed.
Statistical analysis
Comparisons among cells expressing sense FPGS, antisense FPGS, and endogenous FPGS were determined using one way analysis of variance. Tukey’s honestly significance difference test was used for pairwise comparisons. For the in vitro chemosensitivity analyses, plots of percentage of survival versus dose demonstrated S shaped curves, and therefore the logit transformation [logit(p) = ln(p/[1−p])] was used. Ordinary least squares regression was used to model the effect of log(dose) of chemotherapy and cell type (sense FPGS, endogenous FPGS, and antisense FPGS) on the logit transformed proportion of cells that survived at each dose. The interaction between cell type and log(dose) was included in the model to test the hypothesis that the cell types were differentially sensitive to chemotherapy. IC25, IC50, and IC75 doses and their 95% confidence intervals were calculated on the log scale from the regression results, as described previously,34 and then back transformed to the original scale for reporting. For all analyses, results were considered statistically significant if two tailed p values were <0.05. Analyses were performed using SAS, version 8 (SAS Institute, Cary, North Carolina, USA).
RESULTS
HCT116 cells expressing the sense and antisense FPGS had significantly higher and lower steady state levels of the FPGS protein, respectively, compared with those expressing endogenous FPGS (p<0.001) (fig 1A ▶). HCT116 cells expressing the sense FPGS had an 8.2-fold higher, whereas those expressing the antisense FPGS had a 2.3-fold lower, FPGS activity compared with those expressing endogenous FPGS (p<0.001) (fig 1B ▶). HCT116 cells expressing the sense and antisense FPGS grew faster and slower, respectively, than those expressing endogenous FPGS, as reflected by a significantly decreased and increased doubling time, respectively (32.0 (0.6) hours (sense) v 34.9 (0.4) hours (endogenous) v 36.4 (0.7) hours (antisense); p<0.001).
Figure 1.
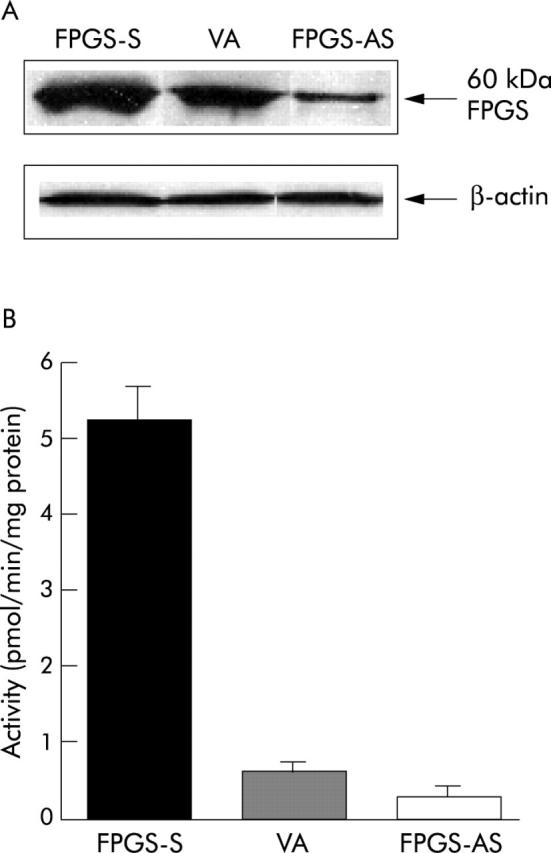
HCT116 colon cancer cells transfected with the sense (FPGS-S) and antisense (FPGS-AS) folylpolyglutamate synthetase (FPGS) cDNA had significantly higher and lower steady state levels of FPGS protein (A) and FPGS activity (B), respectively, compared with cells transfected with vector alone (VA; endogenous FPGS) (p<0.001).
Following conjugase treatment (which allows measurement of total folate content, including short and long chain polyglutamates), intracellular folate concentration of HCT116 cells expressing the sense FPGS was 30% higher, whereas that of those expressing the antisense FPGS was 21% lower, than that of cells expressing endogenous FPGS (p<0.0001) (fig 2 ▶). Intracellular folate concentration of samples not treated with conjugase (which allows determination of short chain polyglutamates) was not significantly different between cells expressing the sense FPGS and those expressing endogenous FPGS and between cells expressing the antisense FPGS and cells expressing endogenous FPGS (fig 2 ▶). However, there were significantly more short chain polyglutamates in cells expressing the sense FPGS compared with those expressing the antisense FPGS (p<0.05) (fig 2 ▶). Differences between mean folate concentration of conjugase treated and untreated samples (which allows determination of long chain polyglutamates) however clearly demonstrate that cells expressing the sense FPGS have significantly higher concentrations of long chain polyglutamates than cells expressing endogenous FPGS and those expressing the antisense FPGS, and that cells expressing the antisense FPGS have significantly lower concentrations of long chain polyglutamates than those expressing endogenous FPGS and cells expressing the sense FPGS (absolute differences of 7.6 for cells expressing the sense FPGS, 5.4 for cells expressing endogenous FPGS, and 3.9 for cells expressing the antisense FPGS; p<0.001) (fig 2 ▶). These observations confirm that the sense and antisense FPGS modulation in this system has appropriately affected the glutamate chain lengths of intracellular folate pool.
Figure 2.

Intracellular folate concentrations of HCT116 cells expressing the sense (FPGS-S), endogenous (vector alone (VA)), and antisense (FPGS-AS) folylpolyglutamate synthetase (FPGS). Conjugase treatment allows measurement of total folate content, including short and long chain polyglutamates, whereas non-conjugase treatment determines the content of short chain polyglutamates. Intracellular concentrations of total folate were significantly higher in cells expressing the sense FPGS and lower in cells expressing the antisense FPGS, respectively, than in those expressing endogenous FPGS (p<0.0001). Differences between mean folate concentration for conjugase treated and untreated samples (which allows determination of long chain polyglutamates) were significantly higher in cells expressing the sense FPGS and lower in cells expressing the antisense FPGS, respectively, compared with those expressing endogenous FPGS (p<0.001).
Cells expressing the sense FPGS had significantly higher, whereas those expressing the antisense FPGS had significantly lower, GGH mRNA expression, compared with those expressing endogenous FPGS (p<0.05) (fig 3 ▶).
Figure 3.

HCT116 cells expressing the sense folylpolyglutamate synthetase (FPGS-S) had significantly higher, whereas those expressing the antisense FPGS (FPGS-AS) had significantly lower, γ-glutamyl hydrolase (GGH) mRNA expression compared with those expressing endogenous FPGS (vector alone (VA)) (p<0.05), as determined by real time quantitative reverse transcription-polymerase chain reaction. GGH is a lysosomal peptidase that removes the terminal glutamates, thereby counterbalancing the action of FPGS. Human 5-aminolevulinate delta synthase 1 was used as the endogenous reference gene (primers: forward, 5′-TGC CCA TTC TTA TCC CGA GT-3′; reverse, 5′-GGT TTC TTT GAT CTG TTG GTA GTG-3′). Relative quantification was performed using LC Relative Quantification Software version 1.0 (Roche Diagnostics).
In vitro chemosensitivity to 5-FU plus LV was significantly different among HCT116 cells transfected with the three different FPGS constructs (p<0.001) (fig 4A ▶). In vitro chemosensitivity of HCT116 cells expressing the sense FPGS to 5-FU plus LV was significantly enhanced compared with those expressing endogenous or the antisense FPGS (p<0.0001) (fig 4A ▶). In contrast, in vitro chemosensitivity of HCT116 cells expressing the antisense FPGS was significantly decreased compared with those expressing endogenous or the sense FPGS (p = 0.0122 and p<0.0001, respectively) (fig 4A ▶). IC25, IC50, and IC75 values for 5-FU indicate significantly enhanced chemosensitivity of HCT116 cells expressing the sense FPGS and reduced chemosensitivity of those expressing the antisense FPGS compared with cells expressing endogenous FPGS (table 1 ▶).
Figure 4.

(A) In vitro chemosensitivity to 5-fluorouracil (5-FU) plus leucovorin (LV) was significantly different among HCT116 colon cancer cells expressing the sense (FPGS-S), endogenous (vector alone (VA)), and antisense (FPGS-AS) folylpolyglutamate synthetase (FPGS) (p<0.001). Chemosensitivity of cells expressing the sense FPGS to 5-FU+LV was significantly enhanced compared with those expressing endogenous or the antisense FPGS (p<0.0001). In contrast, chemosensitivity of cells expressing the antisense FPGS was significantly decreased compared with those expressing endogenous or the sense FPGS (p = 0.0122 and p<0.0001, respectively). (B) In vitro chemosenstivity to methotrexate (MTX) was not significantly different among HCT116 cells expressing the sense, endogenous, and antisense FPGS (p = 0.548).
Table 1.
IC25, IC50, and IC75 values of 5-fluorouracil (5-FU) and methotrexate (MTX) in HCT116 colon cancer cells expressing sense (FPGS-S), endogenous (vector alone (VA)), and antisense (FPGS-AS) folylpolyglutamate synthetase (FPGS)
| 25% survival | 50% survival | 75% survival | ||||
| IC25 | 95% CI | IC50 | 95% CI | IC75 | 95% CI | |
| 5-FU (μM) | ||||||
| FPGS-S | 3.90 | (3.30, 4.63) | 1.64 | (1.26, 2.14) | 0.35 | (0.26, 0.47) |
| VA | 17.03 | (15.39, 18.83) | 3.03 | (2.71, 3.37) | 0.54 | (0.42, 0.68) |
| FPGS-AS | 59.83 | (48.83, 73.30) | 4.57 | (4.15, 5.02) | 0.69 | (0.47, 1.01) |
| MTX (nM) | ||||||
| FPGS-S | 13.93 | (10.99, 17.67) | 9.54 | (8.18, 11.13) | 6.54 | (6.05, 7.07) |
| VA | 13.18 | (12.38, 14.04) | 8.82 | (8.49, 9.16) | 5.89 | (5.79, 6.00) |
| FPGS-AS | 10.95 | (10.32, 11.61) | 7.53 | (7.29, 7.79) | 5.18 | (5.10, 5.27) |
IC25, IC50, and IC75 values (that is, drug concentration that corresponded to a reduction in cell survival by 75%, 50%, and 25%, respectively, compared with the survival of untreated control cells) were calculated from plots of drug concentration versus proportion of cells that survived.
Overall, there was no significant difference in in vitro chemosensitivity to MTX among HCT116 cells transfected with the three different FPGS constructs (p = 0.548) (fig 4B ▶). IC25, IC50, and IC75 values of MTX, however, suggest that antisense FPGS inhibition enhances, whereas sense FPGS overexpression decreases, chemosensitivity of HCT116 cells to MTX compared with cells expressing endogenous FPGS (table 1 ▶).
We wished to ascertain that the observed effects on chemosensitivity of HCT116 cells to 5-FU and MTX were secondary to alterations in FPGS and not an unintended effect of FPGS modulation on intrinsic TS protein expression and activity. TS is a critical target for 5-FU and MTX, and its expression level is an important predictor of chemosensitivity of cancer cells to 5-FU and MTX.35 TS protein expression and catalytic activity were similar among HCT116 cells expressing the sense, antisense, and endogenous FPGS that were not treated with 5-FU or MTX (data not shown). The effect of FPGS modulation on two molecular determinants of chemosensitivity, p53 and p21,36–38 was also investigated. FPGS overexpression downregulated, whereas FPGS inhibition upregulated, p53 and p21 protein expression compared with endogenous FPGS (fig 5 ▶).
Figure 5.
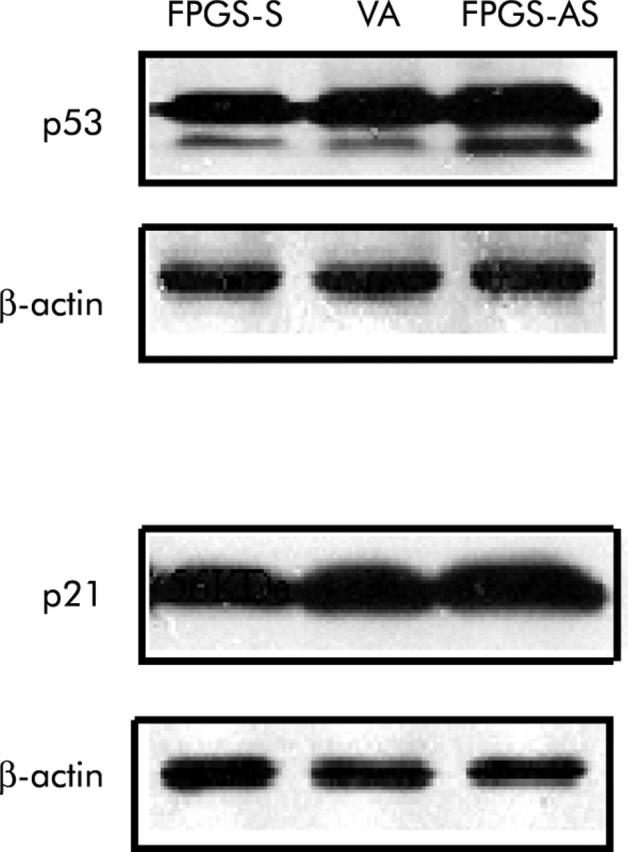
Effect of folylpolyglutamate synthetase (FPGS) modulation on p53 and p21. p53 is integrally involved in cell cycle control, DNA repair, and apoptosis50 while p21 is a member of the cyclin dependent kinase inhibitor family, which inhibits G1 and S phase progression.51 p53 promotes cell cycle arrest in late G1 through upregulation of p21 in response to cell injury, thereby allowing time for repair, whereas in the event that DNA damage is more severe and non-reparable, p53 performs its alternative role of moving the cell into apoptosis.50 FPGS overexpression (FPGS-S) downregulated, whereas FPGS inhibition (FPGS-AS) upregulated, p53 and p21 protein expression compared with endogenous FPGS (vector alone (VA)) in HCT116 cells (densitometry of bands: p53:β-actin ratio 1.9 (FPGS-S) v 3.1 (VA) v 4.1 (FPGS-AS); p21:β-actin ratio 1.7 (FPGS-S) v 2.5 (VA) v 3.2 (FPGS-AS)).
DISCUSSION
We developed an in vitro model of FPGS overexpression and inhibition in HCT116 colon cancer cells with predictable functional consequences. Compared with HCT116 cells expressing endogenous FPGS, those expressing the sense FPGS had significantly higher FPGS expression and activity, higher GGH expression, faster growth rates, higher concentrations of total intracellular folate, and higher content of long chain polyglutamates. In contrast, HCT116 cells expressing the antisense FPGS had significantly lower FPGS expression and activity, lower GGH expression, slower growth rates, lower concentrations of total intracellular folate, and lower content of long chain polyglutamates compared with those expressing endogenous FPGS. These observed metabolic and functional consequences of FPGS overexpression and inhibition are consistent with the known biological function of FPGS and provided an appropriate in vitro model to test the effect of FPGS overexpression and inhibition on chemosensitivity of colon cancer cells to 5-FU and MTX.
Using this system, we have shown that FPGS overexpression enhances, whereas FPGS inhibition decreases, chemosensitivity of colon cancer cells to 5-FU plus LV. The most likely explanation for this observation is that FPGS overexpression enhanced the cytotoxic effect of 5-FU by increasing relative intracellular concentrations of longer chain length 5,10-methyleneTHF polyglutamates, resulting in more efficient formation and stabilisation of the inhibitory 5,10-methyleneTHF-TS-FdUMP ternary complex. In contrast, FPGS inhibition decreased the cytotoxic effect of 5-FU by decreasing relative intracellular concentrations of longer chain length 5,10-methyleneTHF polyglutamates, resulting in less efficient formation and stabilisation of the 5,10-methyleneTHF-TS-FdUMP ternary complex. However, we did not provide direct evidence that FPGS modulation lead to different glutamate chain lengths of 5,10-methyleneTHF because of technical difficulties in measuring intracellular 5,10-methyleneTHF. Furthermore, we did not determine and compare the rate of formation and dissociation and the concentration of the 5,10-methyleneTHF-TS-FdUMP ternary complex among HCT116 cells transfected with the three different FPGS constructs. However, indirect evidence from a prior study23 supports the proposed mechanism by which FPGS modulation might have affected chemosensitivity of colon cancer cells to 5-FU. In that study, the effect of LV, which is converted to 5,10-methyleneTHF and enters the folate pathway,21 on chemosensitivity of human leukaemia CCRF-CEM (the parent cell line with proficient polyglutamylation) and CCRF-CEM/P (a cell line with impaired ability to form polyglutamates) to 5-FU was compared.23 Both CCRF-CEM and CCRF-CEM/P cells accumulated 5,10-methyleneTHF in the presence of LV in a dose dependent manner.23 However, at a dose of 5-FU that produced only a slight decrease in cell growth, addition of LV further inhibited the cell growth in CCRF-CEM cells, but not in CCRF-CEM/P cells,23 suggesting that the impaired polyglutamylation of 5,10-methyleneTHF was likely responsible for the lack of potentiation of the cytotoxic effect of 5-FU by LV in CCRF-CEM/P cells.
The observed effect of FPGS modulation on chemosenstivity to 5-FU is consistent with the existing in vitro evidence that suggests that the cytotoxic effect of 5-FU is directly correlated with FPGS activity in 14 human cancer cell lines.25 Another in vitro study has shown that human colon cancer HCT8 cells become rapidly resistant to 5-FU over time owing to a progressive decrease in FPGS mRNA expression and activity.24 In contrast, three small human studies have shown conflicting results concerning the role of FPGS activity and mRNA expression in predicting treatment response to 5-FU+LV and survival in patients with colon cancer.39–41 Large clinical trials are therefore necessary to investigate the potential clinical utility of FPGS status as a predictor of treatment response and a prognostic indicator in colon cancer patients receiving 5-FU based chemotherapy.
FPGS modulation had no significant overall effect on the chemosensitivity of colon cancer cells to MTX, in contrast with our hypothesis that FPGS induced changes in the glutamate chain lengths of MTX would affect chemosensitivity of colon cancer cells to MTX by altering intracellular MTX retention and affinity of MTX for its target folate dependent enzymes in the thymidylate and purine biosynthetic pathways. However, the overall pattern of chemosensitivity to MTX and the IC25, IC50, and IC75 values of MTX suggest that FPGS inhibition may enhance, whereas FPGS overexpression may decrease, chemosensitivity of colon cancer cells to MTX. Our data contrast with prior observations, which suggested that FPGS downregulation leads to resistance, whereas FPGS overexpression enhances chemosensitivity, to MTX and novel antifolates in other cancer cell lines.12–18 It is possible that our results are specific to the cell type studied because there are tissue specific differences in MTX metabolism and in FPGS expression and activity.1,2 Also, colon cancer cells are generally not sensitive to MTX.42 Another possible explanation is that the effect of the FPGS induced changes in the glutamate chain lengths of MTX might have been nullified by similar changes in the glutamate chain lengths of intracellular folate induced by FPGS modulation.43–47
We have found that FPGS overexpression downregulates, whereas FPGS inhibition upregulates, p53 and p21 expression in untreated HCT116 cells. Whether or not FPGS modulation induced p53 and p21 changes played a role in altering chemosensitivity of HCT116 cells to 5-FU was not determined in the present study. Given the very small magnitude of changes in p53 and p21 protein expression, however, it is highly unlikely that they contributed significantly to altered chemosensitivity. Nevertheless, the direction of changes of p53 and p21 is consistent with the observed alterations in intracellular folate concentrations resulting from FPGS modulation (that is, the effect of intracellular folate level on DNA damage and consequent p53 activation and p21 upregulation48,49).
At present, mechanisms by which FPGS expression and activity are decreased or qualitatively altered in cancer cells resistant to MTX and 5-FU12–18,23–25 are not clearly elucidated. To date, only one putative mutation in FPGS has been identified.15 Therefore, it is of great interest to elucidate genetic, epigenetic, and other potential mechanisms of FPGS regulation in tumours.
In conclusion, our study provides evidence that FPGS overexpression enhances, whereas FPGS inhibition decreases, chemosensitivity of colon cancer cells to 5-FU. Our data suggest that FPGS status may be an important determinant, and hence a useful clinical predictor, of the chemosensitivity of colon cancer cells to 5-FU based chemotherapy. Furthermore, our data provide, for the first time, evidence suggesting that FPGS gene transfer may be a potential target for increasing sensitivity of colon cancer cells to 5-FU based chemotherapy. In contrast, FPGS modulation appears to have no significant effect on the chemosensitivity of colon cancer cells to MTX. However, our data based on a single colon cancer cell line need to be confirmed in other colon cancer cell lines.
Acknowledgments
This project has been supported by an operating grant (grant #14126) and New Investigator award from the Canadian Institutes of Health Research (to YIK).
Abbreviations
AS, antisense
FdUMP, 5-fluoro-2-deoxyuridine-5-monophosphate
FPGS, folylpolyglutamate synthetase
5-FU, 5-fluorouracil
GGH, γ-glutamyl hydrolase
LV, leucovorin
MTX, methotrexate
OD, optical density
S, sense
THF, tetrahydrofolate
TS, thymidylate synthase
VA, vector alone
APPENDIX
The dose range of 5-FU (1.5–25 μM) used in the present study is clinically relevant based on the pharmacokinetics of 5-FU in humans. After intravenous injection of conventional bolus doses of 400–600 mg/m2 (10–15 mg/kg), peak plasma 5-FU concentrations reach 0.1–1.0 mM.52 Thereafter, rapid metabolic elimination leads to a fall in plasma levels with a primary half life of 6–20 minutes (approximately 10 μM at 60 minutes).52 Plasma 5-FU levels above 1 μM, thought to be the approximate threshold for exerting cytotoxic effects on normal tissues, are maintained for only a few hours.52 Continuous intravenous infusion of 5-FU (1250–2250 mg/m2/day) produces steady state plasma levels of 0.5–7.5 μM.52 The observed IC50 values for 5-FU in cells transfected with the three different FPGS constructs in the present study (1.64–4.57 μM) fall within this range. Also, the observed IC25 value for 5-FU in cells overexpressing FPGS in the present study was 3.90 μM. Previous in vitro studies testing the chemosensitivity of colon cancer cell lines to 5-FU used 5-FU concentrations ranging from 10−4 to 10−10 M with IC50 values for 5-FU ranging from 0.5 to 7.0 μM.53 In contrast, colon cancer cells are generally not sensitive to MTX, and MTX is not routinely used in the treatment of colon cancer.42 In most culture systems, MTX exhibits an IC50 in the 1–10 nM range,54 similar to the observed IC50 values for MTX in the present study.
We did not determine the exact glutamate chain lengths of each intracellular folate derivatives. However, the microbiological technique used in the present study has previously been validated as a good screening tool to determine overall pool of long chain versus short chain polyglutamates.30 The normal intracellular folates are penta-glutamates. It is not known whether FPGS overexpression lead to a higher proportion of penta-glutamates or longer (>5) glutamate chain lengths than cells expressing endogenous or the antisense FPGS in the present study, and hence further studies are required to clarify this issue.
Presented in part at the 2003 American Gastroenterological Association meeting, May 17–22, 2003, Orlando, FL, and published in abstract form in Gastroenterology 2003;124(suppl 1):A238.
REFERENCES
- 1.Shane B . Folate chemistry and metabolism. In: Bailey LB, ed. Folate in health and disease. New York: Marcel Dekker, 1995:1–22.
- 2.Moran RG. Roles of folylpoly-gamma-glutamate synthetase in therapeutics with tetrahydrofolate antimetabolites: an overview. Semin Oncol 1999;26:24–32. [PubMed] [Google Scholar]
- 3.Kamen B . Folate and antifolate pharmacology. Semin Oncol 1997;24:S18–39. [PubMed] [Google Scholar]
- 4.Balinska M , Galivan J, Coward JK. Efflux of methotrexate and its polyglutamate derivatives from hepatic cells in vitro. Cancer Res 1981;41:2751–6. [PubMed] [Google Scholar]
- 5.Jolivet J , Schilsky RL, Bailey BD, et al. Synthesis, retention, and biological activity of methotrexate polyglutamates in cultured human breast cancer cells. J Clin Invest 1982;70:351–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samuels LL, Moccio DM, Sirotnak FM. Similar differential for total polyglutamylation and cytotoxicity among various folate analogues in human and murine tumor cells in vitro. Cancer Res 1985;45:1488–95. [PubMed] [Google Scholar]
- 7.Allegra CJ, Chabner BA, Drake JC, et al. Enhanced inhibition of thymidylate synthase by methotrexate polyglutamates. J Biol Chem 1985;260:9720–6. [PubMed] [Google Scholar]
- 8.Allegra CJ, Hoang K, Yeh GC, et al. Evidence for direct inhibition of de novo purine synthesis in human MCF-7 breast cells as a principal mode of metabolic inhibition by methotrexate. J Biol Chem 1987;262:13520–6. [PubMed] [Google Scholar]
- 9.Moran RG, Colman PD, Jones TR. Relative substrate activities of structurally related pteridine, quinazoline, and pyrimidine analogs for mouse liver folylpolyglutamate synthetase. Mol Pharmacol 1989;36:736–43. [PubMed] [Google Scholar]
- 10.Baldwin SW, Tse A, Gossett LS, et al. Structural features of 5,10-dideaza-5,6,7,8-tetrahydrofolate that determine inhibition of mammalian glycinamide ribonucleotide formyltransferase. Biochemistry 1991;30:1997–2006. [DOI] [PubMed] [Google Scholar]
- 11.Allegra CJ, Drake JC, Jolivet J, et al. Inhibition of phosphoribosylaminoimidazolecarboxamide transformylase by methotrexate and dihydrofolic acid polyglutamates. Proc Natl Acad Sci U S A 1985;82:4881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pizzorno G , Mini E, Coronnello M, et al. Impaired polyglutamylation of methotrexate as a cause of resistance in CCRF-CEM cells after short-term, high-dose treatment with this drug. Cancer Res 1988;48:2149–55. [PubMed] [Google Scholar]
- 13.McCloskey DE, McGuire JJ, Russell CA, et al. Decreased folylpolyglutamate synthetase activity as a mechanism of methotrexate resistance in CCRF-CEM human leukemia sublines. J Biol Chem 1991;266:6181–7. [PubMed] [Google Scholar]
- 14.Mauritz R , Peters GJ, Priest DG, et al. Multiple mechanisms of resistance to methotrexate and novel antifolates in human CCRF-CEM leukemia cells and their implications for folate homeostasis. Biochem Pharmacol 2002;63:105–15. [DOI] [PubMed] [Google Scholar]
- 15.Liani E , Rothem L, Bunni MA, et al. Loss of folylpoly-gamma-glutamate synthetase activity is a dominant mechanism of resistance to polyglutamylation-dependent novel antifolates in multiple human leukemia sublines. Int J Cancer 2003;103:587–99. [DOI] [PubMed] [Google Scholar]
- 16.Sanghani SP, Sanghani PC, Moran RG. Identification of three key active site residues in the C-terminal domain of human recombinant folylpoly-gamma-glutamate synthetase by site-directed mutagenesis. J Biol Chem 1999;274:27018–27. [DOI] [PubMed] [Google Scholar]
- 17.Zhao R , Titus S, Gao F, et al. Molecular analysis of murine leukemia cell lines resistant to 5,10-dideazatetrahydrofolate identifies several amino acids critical to the function of folylpolyglutamate synthetase. J Biol Chem 2000;275:26599–606. [DOI] [PubMed] [Google Scholar]
- 18.Roy K , Egan MG, Sirlin S, et al. Posttranscriptionally mediated decreases in folylpolyglutamate synthetase gene expression in some folate analogue-resistant variants of the L1210 cell. Evidence for an altered cognate mRNA in the variants affecting the rate of de novo synthesis of the enzyme. J Biol Chem 1997;272:6903–8. [DOI] [PubMed] [Google Scholar]
- 19.Kim JS, Lowe KE, Shane B. Regulation of folate and one-carbon metabolism in mammalian cells. IV. Role of folylpoly-gamma-glutamate synthetase in methotrexate metabolism and cytotoxicity. J Biol Chem 1993;268:21680–5. [PubMed] [Google Scholar]
- 20.Aghi M , Kramm CM, Breakefield XO. Folylpolyglutamyl synthetase gene transfer and glioma antifolate sensitivity in culture and in vivo. J Natl Cancer Inst 1999;91:1233–41. [DOI] [PubMed] [Google Scholar]
- 21.Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer 2003;3:330–8. [DOI] [PubMed] [Google Scholar]
- 22.Radparvar S , Houghton PJ, Houghton JA. Effect of polyglutamylation of 5,10-methylenetetrahydrofolate on the binding of 5-fluoro-2′-deoxyuridylate to thymidylate synthase purified from a human colon adenocarcinoma xenograft. Biochem Pharmacol 1989;38:335–42. [DOI] [PubMed] [Google Scholar]
- 23.Romanini A , Lin JT, Niedzwiecki D, et al. Role of folylpolyglutamates in biochemical modulation of fluoropyrimidines by leucovorin. Cancer Res 1991;51:789–93. [PubMed] [Google Scholar]
- 24.Wang FS, Aschele C, Sobrero A, et al. Decreased folylpolyglutamate synthetase expression: a novel mechanism of fluorouracil resistance. Cancer Res 1993;53:3677–80. [PubMed] [Google Scholar]
- 25.Cheradame S , Etienne MC, Chazal M, et al. Relevance of tumoral folylpolyglutamate synthetase and reduced folates for optimal 5-fluorouracil efficacy: experimental data. Eur J Cancer 1997;33:950–9. [DOI] [PubMed] [Google Scholar]
- 26.Sohn KJ, Croxford R, Yates Z, et al. Effect of the methylenetetrahydrofolate reductase C677T polymorphism on chemosensitivity of colon and breast cancer cells to 5-fluorouracil and methotrexate. J Natl Cancer Inst 2004;96:134–44. [DOI] [PubMed] [Google Scholar]
- 27.McGuire JJ, Russell CA. Folylpolyglutamate synthetase expression in antifolate-sensitive and -resistant human cell lines. Oncol Res 1998;10:193–200. [PubMed] [Google Scholar]
- 28.Peters GJ, Laurensse E, Leyva A, et al. Purine nucleosides as cell-specific modulators of 5-fluorouracil metabolism and cytotoxicity. Eur J Cancer Clin Oncol 1987;23:1869–81. [DOI] [PubMed] [Google Scholar]
- 29.Skehan P , Storeng R, Scudiero D, et al. New colorimetric cytotoxicity assay for anticancer-drug screening. J Natl Cancer Inst 1990;82:1107–12. [DOI] [PubMed] [Google Scholar]
- 30.O’Connor DL, Tamura T, Picciano MF. Pteroylpolyglutamates in human milk. Am J Clin Nutr 1991;53:930–4. [DOI] [PubMed] [Google Scholar]
- 31.Yao R , Schneider E, Ryan TJ, et al. Human gamma-glutamyl hydrolase: cloning and characterization of the enzyme expressed in vitro. Proc Natl Acad Sci U S A 1996;93:10134–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cole PD, Kamen BA, Gorlick R, et al. Effects of overexpression of gamma-glutamyl hydrolase on methotrexate metabolism and resistance. Cancer Res 2001;61:4599–604. [PubMed] [Google Scholar]
- 33.Keepers YP, Pizao PE, Peters GJ, et al. Comparison of the sulforhodamine B protein and tetrazolium (MTT) assays for in vitro chemosensitivity testing. Eur J Cancer 1991;27:897–900. [DOI] [PubMed] [Google Scholar]
- 34.Draper NR, Smith H. Applied regression analysis, 2nd Edn. New York: John Wiley & Sons 1981.
- 35.Nagasubramanian R , Innocenti F, Ratain MJ. Pharmacogenetics in cancer treatment. Annu Rev Med 2003;54:437–52. [DOI] [PubMed] [Google Scholar]
- 36.Elsaleh H , Powell B, McCaul K, et al. P53 alteration and microsatellite instability have predictive value for survival benefit from chemotherapy in stage III colorectal carcinoma. Clin Cancer Res 2001;7:1343–9. [PubMed] [Google Scholar]
- 37.Ahnen DJ, Feigl P, Quan G, et al. Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study. Cancer Res 1998;58:1149–58. [PubMed] [Google Scholar]
- 38.Paradiso A , Simone G, Petroni S, et al. Thymidilate synthase and p53 primary tumour expression as predictive factors for advanced colorectal cancer patients. Br J Cancer 2000;82:560–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chazal M , Cheradame S, Formento JL, et al. Decreased folylpolyglutamate synthetase activity in tumors resistant to fluorouracil-folinic acid treatment: clinical data. Clin Cancer Res 1997;3:553–7. [PubMed] [Google Scholar]
- 40.Mini E , Biondi C, Morganti M, et al. Marked variation of thymidylate synthase and folylpolyglutamate synthetase gene expression in human colorectal tumors. Oncol Res 1999;11:437–45. [PubMed] [Google Scholar]
- 41.Odin E , Wettergren Y, Nilsson S, et al. Altered gene expression of folate enzymes in adjacent mucosa is associated with outcome of colorectal cancer patients. Clin Cancer Res 2003;9:6012–9. [PubMed] [Google Scholar]
- 42.Kemeny N , Lokich JJ, Anderson N, et al. Recent advances in the treatment of advanced colorectal cancer. Cancer 1993;71:9–18. [DOI] [PubMed] [Google Scholar]
- 43.Jansen G , Barr H, Kathmann I, et al. Multiple mechanisms of resistance to polyglutamatable and lipophilic antifolates in mammalian cells: role of increased folylpolyglutamylation, expanded folate pools, and intralysosomal drug sequestration. Mol Pharmacol 1999;55:761–9. [PubMed] [Google Scholar]
- 44.Jansen G , Mauritz R, Drori S, et al. A structurally altered human reduced folate carrier with increased folic acid transport mediates a novel mechanism of antifolate resistance. J Biol Chem 1998;273:30189–98. [DOI] [PubMed] [Google Scholar]
- 45.Tse A , Moran RG. Cellular folates prevent polyglutamation of 5, 10-dideazatetrahydrofolate. A novel mechanism of resistance to folate antimetabolites. J Biol Chem 1998;273:25944–52. [DOI] [PubMed] [Google Scholar]
- 46.Peters GJ, Smitskamp-Wilms E, Smid K, et al. Determinants of activity of the antifolate thymidylate synthase inhibitors Tomudex (ZD1694) and GW1843U89 against mono- and multilayered colon cancer cell lines under folate-restricted conditions. Cancer Res 1999;59:5529–35. [PubMed] [Google Scholar]
- 47.O’Dwyer PJ, Nelson K, Thornton DE. Overview of phase II trials of MTA in solid tumors. Semin Oncol 1999;26:99–104. [PubMed] [Google Scholar]
- 48.Ames BN. DNA damage from micronutrient deficiencies is likely to be a major cause of cancer. Mutat Res 2001;475:7–20. [DOI] [PubMed] [Google Scholar]
- 49.Fenech M . The role of folic acid and Vitamin B12 in genomic stability of human cells. Mutat Res 2001;475:57–67. [DOI] [PubMed] [Google Scholar]
- 50.Harris CC. Structure and function of the p53 tumor suppressor gene: clues for rational cancer therapeutic strategies. J Natl Cancer Inst 1996;88:1442–55. [DOI] [PubMed] [Google Scholar]
- 51.Fournel M , Sapieha P, Beaulieu N, et al. Down-regulation of human DNA-(cytosine-5) methyltransferase induces cell cycle regulators p16(ink4A) and p21(WAF/Cip1) by distinct mechanisms. J Biol Chem 1999;274:24250–6. [DOI] [PubMed] [Google Scholar]
- 52.Grem JL. Fluorinated pyrimidines. In: Grem JL, Collins J, eds. Cancer chemotherapy: principles and practices. New York: J B Lippincott Company, 1990:180–224.
- 53.Backus HH, Pinedo HM, Wouters D, et al. Folate depletion increases sensitivity of solid tumor cell lines to 5-fluorouracil and antifolates. Int J Cancer 2000;87:771–8. [DOI] [PubMed] [Google Scholar]
- 54.Preist DG, Bunni MA. Folates and folate antagonists in cancer chemotherapy. In: Bailey LB, ed. Folate in health and disease. New York: Marcel Dekker, 1995:379–403.