

Abstract
Background and aims: The liver is the central organ for the detoxification of numerous xenobiotics, including carcinogens. We studied the influence of cholestasis and biliary cirrhosis on the detoxification, elimination, and tissue distribution of a model compound and food derived carcinogen, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP).
Methods: Wistar rats were injected with 14C-PhIP into the portal vein one or six weeks after common bile duct ligation (CBDL). Bile flow was reconstituted, bile and urine were collected over 120 minutes, and metabolites were analysed using high performance liquid chromatograpy. Total tissue radioactivity levels in several organs as well as tissue bound (ethanol insoluble tissue fraction) radioactivity levels were determined.
Results: Significant downregulation of the transport proteins multidrug resistance associated protein 2 and breast cancer resistance protein was observed in biliary cirrhosis. Biliary excretion of radioactivity was significantly reduced in cholestasis and biliary cirrhosis compared with controls (15 (2.9)% and 3.2 (1)% of the dose v 36.5 (2)%, respectively). Phase II metabolism was severely reduced in cirrhotic rats, resulting in a twofold increase in tissue radioactivity levels in the liver, kidney, and colon. Biliary cirrhosis increased tissue binding of reactive metabolites, as expressed in cpm/100 mg tissue in the liver and the colon (3267 (1218) v 1191 (429) in the liver, 3044 (1913) v 453 (253) in the colon).
Conclusions: Biliary cirrhosis induced by CBDL causes impaired metabolism and elimination of PhIP, and leads to higher tissue levels of potentially genotoxic metabolites in the liver and colon of rats. These data may explain the increased incidence of hepatic and extrahepatic cancers in cholestasis and liver cirrhosis.
Keywords: biliary cirrhosis, biliary secretion, carcinogenesis, hepatic metabolism
Epidemiological studies have shown that cirrhosis is associated with an increased risk of hepatocellular carcinoma.1 The effect of cirrhosis on the incidence of other cancers is unclear but studies suggest an increased risk of cancers in the respiratory and gastrointestinal tracts, as well as urinary organs.2 Primary sclerosing cholangitis (PSC), a chronic cholestatic disease, is known to be a risk factor for hepatic and extrahepatic malignancies,3 including colon carcinoma in patients with ulcerative colitis.4
Chronic cholestasis leads to downregulation of multidrug resistance associated protein 2 (Mrp2)5; the behaviour of breast cancer resistance protein (Bcrp) during cholestasis is unknown. Protein levels of cytochrome P450 (CYP) isoforms are differentially regulated in biliary cirrhosis and cholestasis: while CYP1A, 2C, and 3A isoforms were reduced at the protein and activity level, levels of CYP2E1 were unchanged.6 Furthermore, protein levels of UDP glucuronosyltransferases (UGT) have been shown to be preserved.7 Therefore, the presence of cirrhosis is likely to lead to impaired metabolism and excretion of substrates of CYP isoforms and of the transporter Mrp2. However, protein levels may not always be representative of enzyme activity, and reduced protein levels may still maintain metabolic fluxes, as in the case of Mrp2.8 Further, in vitro assays measuring isolated activity do not always correctly predict the in vivo behaviour of drugs or toxins. Many xenobiotic compounds are substrates for several different steps in metabolism and detoxification, and some substrates can be metabolised by different isoforms of the same enzyme complex. We have previously shown that the absence of a whole class of metabolising enzymes (in this case, all UGT1A isoforms) does not necessarily lead to impaired metabolism of substrates as there may be upregulation of compensatory mechanisms of redundant enzyme systems such as UGT2 isoforms and sulphotransferases.9 Therefore, it is difficult to predict how liver cirrhosis or cholestasis may affect the overall metabolism and detoxification of toxic compounds.
To determine the effect of cholestasis and cirrhosis on metabolism, detoxification, and elimination of a food derived carcinogen, we studied the fate and distribution of a model substrate and further characterised the molecular alterations in biliary cirrhosis after six weeks of common bile duct ligation (CBDL). To determine primarily changes in liver metabolism and to exclude alterations via intestinal uptake, we administered the model substrate 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) intravenously into the portal vein.
PhIP is a heterocyclic amine and as such is the most abundant food derived carcinogen in prepared meat.10 Its metabolism has been extensively studied in several species,10,11 and we and others have previously shown that PhIP and its major metabolites are substrates of the efflux pump MRP2 in rats and humans.12,13 Additionally, it has been shown recently that the parent compound is transported very efficiently by the breast cancer resistance protein (Bcrp).14 Both of these transport proteins are present in the liver and intestine.15 Activation of PhIP to N-OH-PhIP, a genotoxic compound and precursor for further activation, occurs via CYP1A1 and CYP1A2. This enzymatic reaction can also yield 4′-OH-PhIP, which is the first detoxified metabolite of PhIP. Both hydroxylation products can be further metabolised by glucuronidation16 or sulphation.17 Detoxification of PhIP may also occur via direct glucuronidation of the parent compound, a pathway which has only limited importance in rodents but which may account for approximately 20% of metabolites in humans.18 Data from animal studies have shown that PhIP is involved in rat colon carcinogenesis19 but data regarding the contribution of PhIP to human colon carcinogenesis are controversial (discussed recently by Berlau and colleagues20), which may be due in part to methodological problems.21 In the light of reports on the association between colon cancer and liver cirrhosis,2 we studied the metabolism and excretion of PhIP as a carcinogenic model compound in cholestatic (one week of bile duct ligation) and cirrhotic (six weeks of bile duct ligation) animal models.
MATERIALS AND METHODS
Chemicals
[14C]PhIP (specific activity 10 mCi/mmol) was obtained from Toronto Research Chemicals Inc. (North York, Ontario, Canada). All solvents used for high performance liquid chromatograpy (HPLC) separation (HPLC or analysis grade) were from Merck (Darmstadt, Germany).
Animals
Female Wistar rats (Janvier, Le Genest-St Isle, France), 200–250 g each, were used for all experiments. Rats were housed in cages with a 12 hour light/dark cycle and given access to food and water ad libitum. CBDL was executed as previously described.22 Briefly, after subcutaneous injection of ketamine (80 mg/kg body weight) and xylazine (4 mg/kg body weight), the abdomen was opened through a midline incision. The bile duct was identified, isolated, and double ligated in the proximal part. In sham operated rats, the bile duct was isolated and touched but not ligated. After one and six weeks, respectively, all rats again received ketamine/xylazine, the abdomen was opened, and the fate and distribution of PhIP was studied, as previously described.12 Briefly, the bile duct was cannulated, in CBDL rats proximal to the ligation (to restore bile flow). The urine bladder was also cannulated. Rats received 2 µCi of [14C]PhIP, dissolved in 500 µl of 10 mM tauroursodeoxycholate, intravenously into the portal vein. Bile and urine were collected, rats were sacrificed after two hours by bleeding from the aorta, and the liver, colon, small intestine, pancreas, kidneys, heart, and lungs were harvested. Faeces were separated from the intestine before storage at −80°C. All animal experiments were approved by the local animal care committee.
Analysis of bile and urine samples
Scintillation fluid was added to aliquots from bile and urine samples and total radioactivity of each sample was measured; 20 µl from each bile and urine sample were directly injected onto a HPLC system, as previously described.12 Briefly, a 0.1% diethylamine/methanol gradient was used at pH 6.0 to separate the metabolites on a Gynkotek-HPLC-system (Germering, Germany). Radioactivity was monitored with a Packard 515TR scintillation analyser (Meriden, Connecticut, USA). HPLC chromatograms were used for quantification of each metabolite using identification data from previous experiments.12
Organs
Aliquots (∼100 mg) of each organ (from at least two different sites) were dissolved in Soluene 350 (Packard; Meriden) at 40°C, decolourised with 10% (v/v) H2O2, scintillation fluid was added, and the samples were counted. Other aliquots were used for ethanol extraction of metabolites (for analysis on HPLC), as previously described.12 Differences in radioactivity content before and after ethanol extraction were used to estimate the covalent binding of reactive PhIP metabolites to tissue components.23
Western blotting
Liver microsomal extracts were produced to analyse protein levels of Mrp2 and UGT1A isoforms. Liver tissue samples (∼100 mg) were homogenised in 1 ml of homogenisation buffer (0.25 M sucrose, 10 mM HEPES, pH 7.5, 1 mM EDTA, pH 8). After centrifugation (31 000 rpm at 4°C), the pellet was resuspendend in 80 µl of storage buffer (0.3 M sucrose, 10 mM Hepes, pH 7.5) and stored at −80°C. Both buffers contained a commercial mixture of protease inhibitors at the recommended concentration (Mini Complete, Roche, Basel, Switzerland), 0.5 µg/ml pepstatin A, and 40 µg/ml phenylmethylsulphonylfluoride. Protein concentrations were determined according to Bradford.24 After determination of the linear range (25–200 µg), similar amounts of protein (25 µg) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis25 within three days and probed with the antibodies M2III-6 (for rat Mrp2), BXP-53 (for rat Bcrp, kindly provided by Dr Alfred Schinkel), and HEB7 (for rat UGT1A isoforms26) after transfer to nitrocellulose membranes. Equal loading was confirmed by Ponceau staining.
Statistical analysis
Significance was assumed at p<0.05. Testing was done with ANOVA and post testing with the Bonferroni method. All data represent means (SD) from four animals per group.
RESULTS
Physiological and anatomical parameters
Biliary and urinary flow were not significantly different between groups (fig 1A ▶). Liver mass in cirrhotic and cholestatic animals was significantly increased (21.7 (4.7) g and 11.2 (0.6) g, respectively, v 7.9 (0.9) g in sham operated rats) (fig 1B ▶). After six weeks of bile duct ligation, the livers were firm and showed nodule formation. Microscopically, ductular proliferation and cirrhosis were seen mainly in periportal areas.
Figure 1.
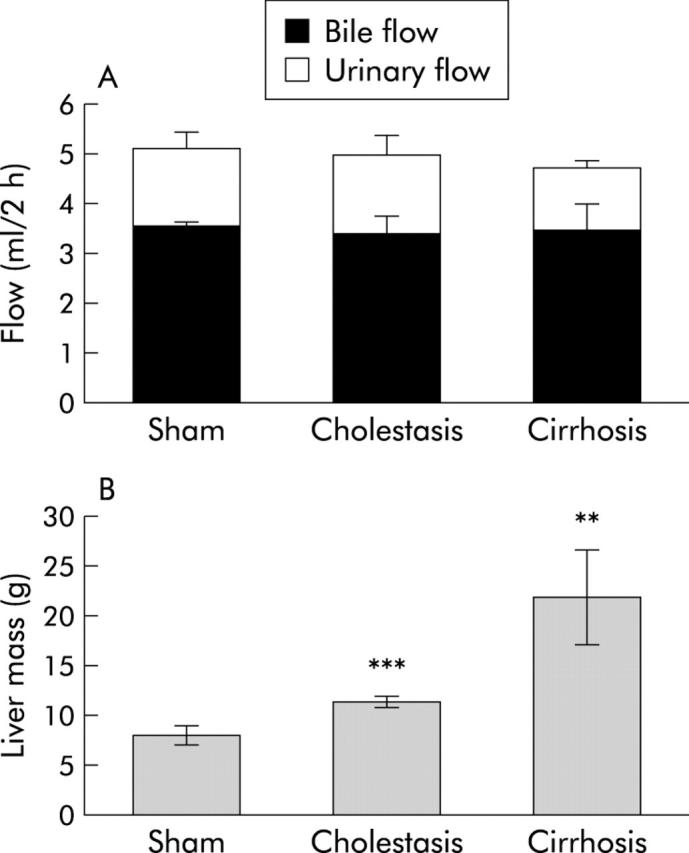
Physiological and anatomical parameters in cholestatic and cirrhotic rats. (A) Bile flow and urinary flow, and (B) liver mass in control, cholestatic, and cirrhotic animals. Significance levels are denoted by **p<0.01 and ***p<0.001.
Protein expression of Mrp2, Bcrp, and UGT1A isoforms in cirrhotic liver after six weeks of bile duct ligation
As the influence of CBDL on Mrp2 expression in rat liver has been investigated only up to 14 days of CBDL, we determined protein levels of Mrp2 in rats with liver cirrhosis after six weeks of CBDL. We confirmed that Mrp2 protein expression decreased further as the duration of bile duct ligation increased, and was ∼10% of sham controls in liver tissue from rats with biliary cirrhosis (p<0.001) (fig 2A ▶). Regulation of Bcrp during CBDL has not been studied previously. We observed a 28% decrease in Bcrp protein to 72 (17)% of sham controls (p = 0.05) (fig 2B ▶). We also confirmed that levels of UGT1A isoforms were unchanged in cirrhotic livers (fig 2C ▶).
Figure 2.

Multidrug resistance associated protein 2 (Mrp2) (A), breast cancer resistance protein (Bcrp) (B) and UDP-glucuronosyltransferase 1A (Ugt1A) (C) protein mass in the livers of cirrhotic rats. Top: representative western blots for two animals in each group; bottom: densitometric analysis (controls = 100%). Significance levels are denoted by *p = 0.05 and ***p<0.001.
Biliary and urinary excretion of radioactive metabolites following injection of 14C-PhIP into the portal vein
Total excretion of radioactivity in two hours was significantly reduced in cholestatic (35.6 (2.8)%) and cirrhotic (9.5 (1.6)%) rats compared with sham operated controls (49 (2.9)%) (fig 3A ▶). Biliary excretion of the radiolabel is shown in fig 3B ▶ and was 3.2 (1)% in cirrhotic, 15 (2.9)% in cholestatic, and 36.5 (2)% in sham operated rats, respectively (see fig 3A ▶). In cholestatic rats, the decrease in biliary excretion was partially compensated for by increased urinary excretion (20.6 (1.9)% v 12.5 (1.7)% in sham operated rats) while in cirrhotic rats urinary excretion was dramatically reduced (6.3 (1)%) (fig 3 ▶).
Figure 3.
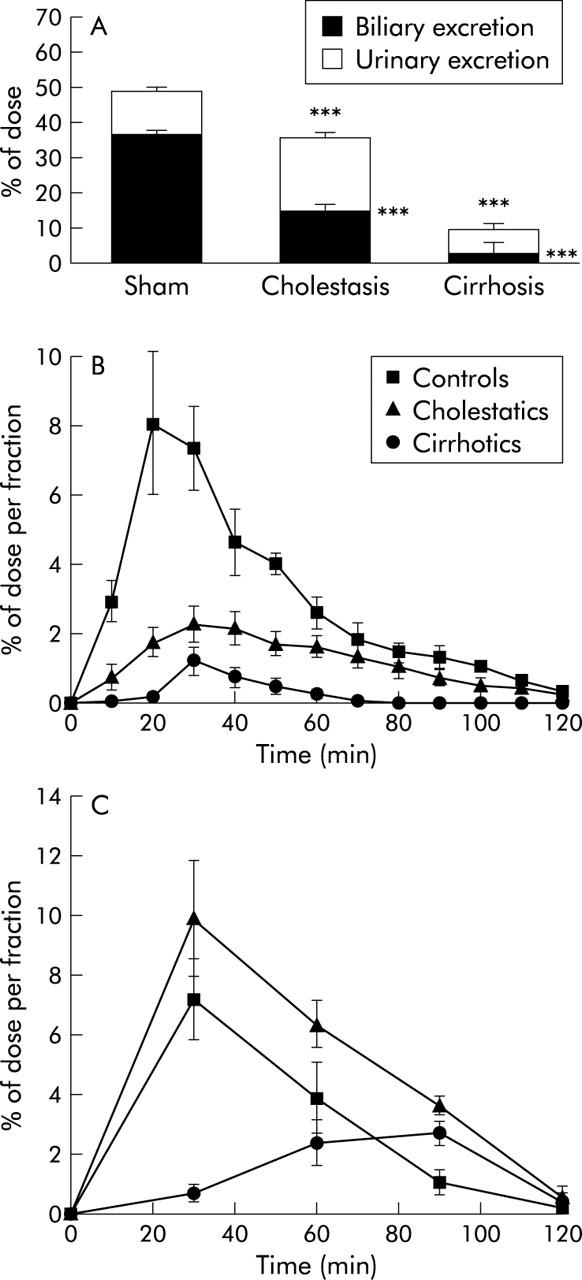
Biliary and urinary excretion of radioactivity. (A) Complete excretion, divided into biliary and urinary excretion. Significance level is denoted by ***p<0.001. Biliary (B) and urinary (C) excretion (fractionated) over time in controls, cholestatic, and cirrhotic rats.
PhIP metabolites in bile, urine and liver
PhIP is metabolised in the body to N-OH- or 4′-OH-PhIP (phase I). Quantitative important phase II metabolites comprise glucuronides of both phase I metabolites (conjugation on the N2-, N3,- or 4′-OH position) as well as sulphation of 4′-OH-PhIP. All of these metabolites were analysed using separation by HPLC.
Biliary excretion of all glucuronidated metabolites was significantly reduced in cholestatic and, more severely, in cirrhotic rats (3.3 (0.2)% and 0.5 (0.2)%, respectively) compared with normal controls (21.1 (1.6)% of injected dose). Biliary excretion of PhIP, the unmetabolised parent compound, was significantly diminished in both animal models to a similar extent. The bile content of PhIP-sulphate was significantly elevated in cholestatic rats (6.8 (1)% v 4.4 (0.3)% in controls) but this compensatory metabolism was absent in cirrhotic rats (0.45 (0.1)%) (fig 4A ▶). In urine, all phase II metabolites were significantly elevated (glucuronides and sulphate) in rats with acute cholestasis compared with controls. This compensation for reduced biliary excretion was again absent in cirrhotic rats (reduced urinary excretion for the N2- and N3-glucuronides and sulphate) (fig 4B ▶). The unmetabolised parent compound PhIP was significantly elevated in the urine of both cholestatic and cirrhotic rats (0.97 (0.16) and 1.35 (0.19)%, respectively, compared with 0.43 (0.1)% in controls) (fig 4B ▶).
Figure 4.
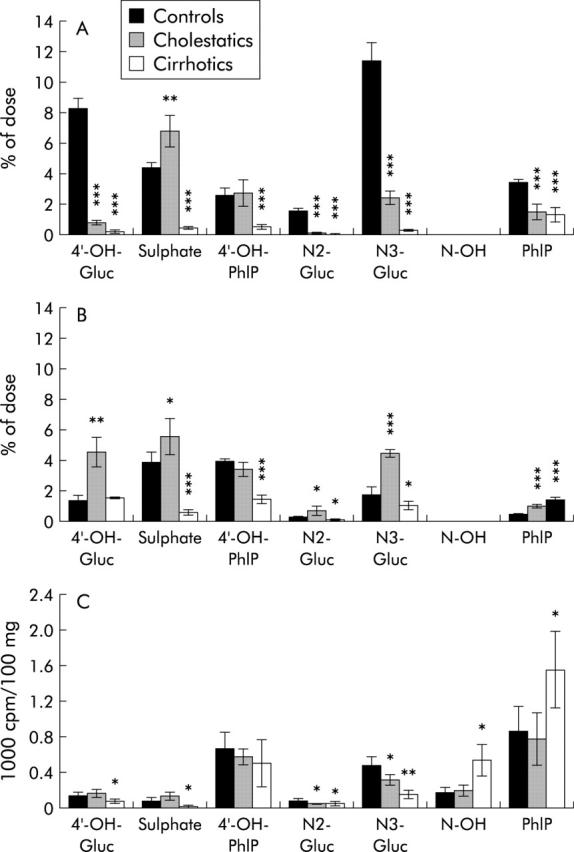
2-Amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) metabolites in (A) bile, (B) urine, and (C) liver tissue. Data for controls, cholestatic, and cirrhotic rats are given as a percentage of the total dose in bile and urine and as cpm/100 mg tissue in liver. Significance levels are denoted by *p<0.05, **p<0.01, ***p<0.001. 4′-OH-Gluc = glucuronides of 4′-OH-PhIP (three different isoforms); N2-/N3-Gluc = glucuronides of N-OH-PhIP (two different isoforms); Sulphate = 4′-sulphate; N-OH = N-OH-PhIP.
In the liver of cirrhotic rats, the content of N-OH-PhIP and PhIP itself was elevated twofold compared with control and cholestatic rats. However, all glucuronides and PhIP-sulphate were significantly reduced (fig 4C ▶).
Blood levels of radioactivity did not differ significantly at two hours after administration in cirrhotic and cholestatic rats compared with sham operated rats (data not shown).
Liver tissue levels of total and bound (ethanol insoluble) radioactivity
In the liver of cirrhotic rats, there was a fourfold accumulation of radioactivity at two hours after intravenous administration compared with control rats (15.9 (1.4)% v 4.5 (1.4)%; p<0.001) (fig 5A ▶). However, when corrected for liver mass, these differences were less apparent (7291 (1906) v 4067 (435) cpm/100 mg tissue; p = 0.02) (fig 5B ▶). A similar effect was observed in cholestatic rats (8.4 (2.6)% radioactivity accumulating in the liver compared with 4.5 (1.4)% in controls; p = 0.02) (fig 5A ▶). However, this difference was not statistically significant when expressed per 100 mg liver mass (4946 (1702) v 4067 (435) cpm/100 g tissue; p = 0.2) (fig 5B ▶). Tissue bound radioactivity was elevated in cirrhotic as well as in cholestatic animal models using both calculation methods (7.1 (2)% and 3.4 (1)%, respectively, v 1.3 (0.4)% percentage of the total dose, p = 0.005 and 0.008, respectively (fig 5C ▶); 3267 (1218) cpm and 2011 (644) cpm, respectively, v 1191 (429) cpm in mass related radioactivity per 100 mg tissue, p = 0.02 and 0.04, respectively) (fig 5D ▶).
Figure 5.

Tissue levels of radioactivity in the liver, kidney and colon. (A) Total radioactivity in whole organs, (B) total radioactivity per 100 mg tissue, (C) tissue bound radioactivity in whole organs, (D) tissue bound radioactivity per 100 mg in controls, cholestatic, and cirrhotic rats. Significance levels are denoted by *p<0.05, **p<0.01, ***p<0.001.
Tissue levels of total and bound (ethanol insoluble) radioactivity in other organs
There were no significant differences in the weight of the kidney, heart, lung, or intestine between the treatment groups. The following results were obtained (see fig 5A ▶–D): accumulation of radioactivity from injected 14C-PhIP was significantly increased in the colon in cirrhotic rats but not in rats with acute cholestasis. In contrast, tissue bound radioactivity was significantly elevated in the colon in both cirrhotic and cholestatic rats compared with control rats (3044 (1913) cpm and 1410 (203) cpm, respectively, v 453 (253) cpm per 100 mg tissue, p = 0.04 and <0.001, respectively). In the kidney, total radioactivity was decreased in cirrhotic compared with cholestatic rats but was significantly elevated in both when compared with control animals (16911 (3987) cpm and 21101 (6248), respectively, v 9809 (2264), p = 0.014 for both). However, tissue bound radioactivity in the kidney was not significantly elevated in cirrhotic or cholestatic rats compared with controls.
Differences in other organs (small intestine, pancreas, heart, lung) were borderline significant, and are not shown.
DISCUSSION
Our data demonstrate that chronic cholestasis and biliary cirrhosis cause impaired hepatic metabolism and excretion of the heterocyclic amine and food derived carcinogen PhIP. Reduced biliary excretion after one week of bile duct ligation could partially be compensated for by increased urinary elimination of PhIP and its metabolites but biliary cirrhosis led to a marked decrease in both elimination routes. One of the consequences of decreased metabolism and elimination was accumulation of PhIP and its metabolites in the liver, kidney, and colon, with the latter being a potential target organ of the carcinogen PhIP in rats. Tissue binding, defined as ethanol insoluble tissue fraction of radioactivity, was significantly increased in the colon of cirrhotic rats, indicating the formation of adducts of activated PhIP metabolites to tissue components, including DNA.
PhIP is a substrate for CYP1A1 and CYP1A2 isoforms, several different UGT isoforms, sulphotransferases, and the transport proteins Mrp2 and Bcrp. As such, its metabolism, tissue distribution, and elimination are influenced in a complex way by progressive cholestasis and biliary cirrhosis where adaptive changes in protein expression and activity at different levels of metabolism and transport take place. We have recently shown that cholestasis downregulates intestinal Mrp2 expression and increases oral bioavailability of PhIP in rats,22 indicating that local processes such as cholestasis may influence gene expression in other organs via systemic mediators.27 The complex metabolism of PhIP with competing pathways and widespread changes in protein activity or expression, not only in the liver, complicates assessment of the present data, even in our experimental setting (portal intravenous administration of the compound, dose higher than in physiological situations to allow quantitation of metabolites). This can give rise to misinterpretation of our data and has to be kept in mind when applying results of the current study to different conditions.
Our data show that under the current conditions, phase I metabolism is preserved despite reduced protein levels of the CYP1A isoforms.6 Glucuronidation is decreased in cholestasis and, more so, in biliary cirrhosis despite the persistence of normal UGT levels (fig 2 ▶). We and others have previously shown that decreased activity of Mrp2 is sufficient to reduce phase II metabolism in liver cells,12,28 and this is probably an effect of product inhibition due to reduced biliary elimination. Downregulation of Mrp2 to 10% of its normal expression may have similar effects on glucuronidation activity. In this study, we have shown for the first time that Bcrp, which secretes the parent compound PhIP into bile, is also downregulated during long term CBDL. This modest downregulation may contribute to the high tissue levels of PhIP observed in the livers of rats with biliary cirrhosis (fig 4C ▶).
As the main step of detoxification, a reduction in glucuronidation may seriously intensify the genotoxicity of PhIP. Increased sulphation as a possible compensatory detoxification pathway was only present in cholestatic but not in cirrhotic rats (fig 4A ▶–C), indicating a further deterioration in metabolism in the cirrhotic liver. Instead of the use of alternative detoxification pathways, overall metabolism in cirrhotic livers was reduced, as shown by the elevated PhIP concentrations, and resulted in a shift to microsomal activation (production of N-OH-PhIP, see fig 4C ▶). N-OH-PhIP is already genotoxic per se but it can be metabolised to N-acetoxy- and N-sulfoxy-PhIP, metabolites which both bind highly to DNA and induce genotoxic effects in vitro and in vivo.29
The metabolism of compounds outside the liver is difficult to quantify. Differences in biliary and urinary metabolite concentrations indicate preserved, or even compensatory increased, glucuronidation in extrahepatic tissues such as the intestine (fig 4 ▶). Redistribution of activated metabolites via the blood to other organs for detoxification has been proposed and should also include N-OH-PhIP.30 As the rat intestine lacks CYP1A2 and CYP1A1 is only present in rat duodenum,31 this mechanism is a prerequisite for UGT mediated detoxification in extrahepatic tissues. However, little is known regarding the influence of liver disease on protein expression levels of extrahepatic metabolising enzymes. It is possible that increased N-OH-PhIP concentrations in the colon not only trigger detoxification via glucuronidation but also provoke further activation by acetylation or sulphation, resulting in increased tissue binding, as observed in our experiments. Clearly, further studies are needed to elucidate these systemic effects in vivo.
Neither biliary nor urinary flow were reduced in cirrhotic rats (fig 1 ▶). Maintenance of bile flow after resolution of six weeks of bile duct obstruction is probably due to the high bile salt pool in cirrhotic livers which compensates for the apparent reduction in bile salt independent bile flow via Mrp2.8 Despite preserved urinary flow, cirrhotic animals were not able to compensate for the loss of biliary excretion by renal elimination (fig 3 ▶). In addition, the observed remaining urinary elimination was delayed (fig 3C ▶). In parallel with this result, we saw reduced total tissue concentrations of PhIP and its metabolites in the kidneys of cirrhotic animals compared with cholestatic animals (fig 5 ▶). It has previously been shown that liver cirrhosis results in reduced renal plasma flow in rats,32 which may explain the reduced and delayed urinary excretion. Reduced hepatic basolateral re-secretion into blood appears to play a minor role as blood levels of radioactivity were unchanged two hours after injection and tissue levels in other organs were elevated (colon) or unchanged.
Tissue levels of PhIP and its metabolites in the liver are not easy to interpret in our animal models due to the steady increase in liver mass with increased duration of bile duct ligation. We measured the absolute increase in radioactivity in cirrhotic livers, which may be the consequence of similar tissue content combined with a three fold enlarged organ (fig 5A ▶). Weight dependent calculations however also showed a relative increase in tissue concentrations (fig 5B ▶). Most critical for potential genotoxic effects is the relative tissue load of tissue bound metabolites. Here we found a threefold increase in the liver of cirrhotic animals but in the colon the respective values were elevated sixfold (fig 5D ▶). Given the fact that PhIP is a rat colon carcinogen, this supports the biological significance of hepatic biliary cirrhosis in the development of colon cancers in rats. However, only binding of reactive PhIP metabolites to DNA of colon stem cells determines the extent of possible carcinogenicity by this heterocyclic amine. With our experimental setup, it is not possible to reliably measure the carcinogenic effect of liver cirrhosis on the colon.
Protein expression levels of metabolising enzymes in human liver fibrosis and cirrhosis have not been systematically investigated. In PSC, MRP2 mRNA was reduced to 27% of healthy controls33 while in advanced primary biliary cirrhosis (stage IV), protein expression levels of MRP2 were decreased,34 indicating similar regulation events as in rats. However, data regarding phase I and phase II enzymes are completely lacking and hence our results cannot be directly extrapolated to the human situation.
In summary, we have shown that biliary cirrhosis can result in elevated tissue binding of activated PhIP metabolites in the colon, the target organ of PhIP carcinogenesis in rats. This is primarily the result of decelerated and reduced metabolism as well as reduced elimination in the liver, but also demonstrates the remote effects of liver disease on other organs such as the kidney and colon. There are substantial differences in PhIP metabolism between rats and humans which has led to the conclusion that PhIP mediated human carcinogenesis is underestimated in rats.35 In humans, higher levels of adducts forming in colon tissue have been observed when directly compared with rats.36 The relation between activation (N-hydroxylation) and detoxification (4′-hydroxylation) is much more unfavourable in humans than in rats (97:1 v 3.3:137). Additionally, the presence of polymorphisms in human genes affecting enzyme activity38 may result in specific subsets of patients with an increased cancer susceptibility in extrahepatic tissue in general, and the colon in particular. In combination, these facts may lead to magnification of the effects seen in the present study in rodents. The extent of PhIP induced carcinogenesis in humans is still debatable but PhIP is only one xenobiotic model compound, representing numerous other genotoxic substances whose metabolism might also be reduced in liver cirrhosis. Systematic epidemiological studies are therefore warranted to study the clinical importance of liver cirrhosis in extrahepatic carcinogenesis.
Acknowledgments
We thank Sabine Beutelspacher for excellent technical assistance, Alfred Schinkel for providing the BXP-53 antibody, and Jane Byrne for critical reading of the manuscript. Supported by the Deutsche Forschungsgemeinschaft (DFG) grant DI 729/3-1 to CGD and SFB 542 TP C1 to AG, SM, and CG.
Abbreviations
Bcrp, breast cancer resistance protein
CBDL, common bile duct ligation
CYP, cytochrome P450
Mrp, multidrug resistance associated protein
PhIP, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine
UGT, UDP-glucuronosyltransferase
PSC, primary sclerosing cholangitis
HPLC, high performance liquid chromatograpy
REFERENCES
- 1.Kuper H , Ye W, Broome U, et al. The risk of liver and bile duct cancer in patients with chronic viral hepatitis, alcoholism, or cirrhosis. Hepatology 2001;34:714–18. [DOI] [PubMed] [Google Scholar]
- 2.Sorensen HT, Friis S, Olsen JH, et al. Risk of liver and other types of cancer in patients with cirrhosis: a nationwide cohort study in Denmark. Hepatology 1998;28:921–5. [DOI] [PubMed] [Google Scholar]
- 3.Bergquist A , Ekbom A, Olsson R, et al. Hepatic and extrahepatic malignancies in primary sclerosing cholangitis. J Hepatol 2002;36:321–7. [DOI] [PubMed] [Google Scholar]
- 4.Broome U , Lofberg R, Veress B, et al. Primary sclerosing cholangitis and ulcerative colitis: evidence for increased neoplastic potential. Hepatology 1995;22:1404–8. [DOI] [PubMed] [Google Scholar]
- 5.Lee J , Boyer JL. Molecular alterations in hepatocyte transport mechanisms in acquired cholestatic liver disorders. Semin Liver Dis 2000;20:373–84. [DOI] [PubMed] [Google Scholar]
- 6.Bastien MC, Leblond F, Pichette V, et al. Differential alteration of cytochrome P450 isoenzymes in two experimental models of cirrhosis. Can J Physiol Pharmacol 2000;78:912–9. [PubMed] [Google Scholar]
- 7.Debinski HS, Mackenzie PI, Lee CS, et al. UDP glucuronosyltransferase in the cirrhotic rat liver. J Gastroenterol Hepatol 1996;11:373–9. [DOI] [PubMed] [Google Scholar]
- 8.Paulusma CC, Kothe MJ, Bakker CT, et al. Zonal down-regulation and redistribution of the multidrug resistance protein 2 during bile duct ligation in rat liver. Hepatology 2000;31:684–93. [DOI] [PubMed] [Google Scholar]
- 9.Dietrich CG, Ottenhoff R, de Waart DR, et al. Lack of UGT1 isoforms in Gunn rats changes metabolic ratio and facilitates excretion of the food-derived carcinogen 2-amino-1-methyl-6- phenylimidazo[4,5-b]pyridine. Toxicol Appl Pharmacol 2001;170:137–43. [DOI] [PubMed] [Google Scholar]
- 10.Eisenbrand G , Tang W. Food-borne heterocyclic amines. Chemistry, formation, occurrence and biological activities. A literature review. Toxicology 1993;84:1–82. [DOI] [PubMed] [Google Scholar]
- 11.Crofts FG, Sutter TR, Strickland PT. Metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by human cytochrome P4501A1, P4501A2 and P4501B1. Carcinogenesis 1998;19:1969–73. [DOI] [PubMed] [Google Scholar]
- 12.Dietrich CG, de Waart DR, Ottenhoff R, et al. Mrp2-deficiency in the rat impairs biliary and intestinal excretion and influences metabolism and disposition of the food-derived carcinogen 2- amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP). Carcinogenesis 2001;22:805–11. [DOI] [PubMed] [Google Scholar]
- 13.Dietrich CG, de Waart DR, Ottenhoff R, et al. Increased bioavailability of the food-derived carcinogen 2-amino-1- methyl-6-phenylimidazo[4,5-b]pyridine in MRP2-deficient rats. Mol Pharmacol 2001;59:974–80. [DOI] [PubMed] [Google Scholar]
- 14.van Herwaarden AE, Jonker JW, Wagenaar E, et al. The breast cancer resistance protein (Bcrp1/Abcg2) restricts exposure to the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Res 2003;63:6447–52. [PubMed] [Google Scholar]
- 15.Dietrich CG, Geier A, Oude Elferink RP. ABC of oral bioavailability: transporters as gatekeepers in the gut. Gut 2003;52:1788–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nowell SA, Massengill JS, Williams S, et al. Glucuronidation of 2-hydroxyamino-1-methyl-6-phenylimidazo[4, 5- b]pyridine by human microsomal UDP-glucuronosyltransferases: identification of specific UGT1A family isoforms involved. Carcinogenesis 1999;20:1107–14. [DOI] [PubMed] [Google Scholar]
- 17.Lang NP, Nowell S, Malfatti MA, et al. In vivo human metabolism of [2-14C]2-amino-1-methyl-6-phenylimidazo[4,5- b]pyridine (PhIP). Cancer Lett 1999;143:135–8. [DOI] [PubMed] [Google Scholar]
- 18.Langouet S , Paehler A, Welti DH, et al. Differential metabolism of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine in rat and human hepatocytes. Carcinogenesis 2002;23:115–22. [DOI] [PubMed] [Google Scholar]
- 19.Ito N , Hasegawa R, Sano M, et al. A new colon and mammary carcinogen in cooked food, 2-amino-1-methyl-6- phenylimidazo[4,5-b]pyridine (PhIP). Carcinogenesis 1991;12:1503–6. [DOI] [PubMed] [Google Scholar]
- 20.Berlau J , Glei M, Pool-Zobel BL. Colon cancer risk factors from nutrition. Anal Bioanal Chem 2004;378:737–43. [DOI] [PubMed] [Google Scholar]
- 21.Sinha R . An epidemiologic approach to studying heterocyclic amines. Mutat Res 2002;506–507:197–204. [DOI] [PubMed] [Google Scholar]
- 22.Dietrich CG, Geier A, Salein N, et al. Consequences of bile-duct obstruction on intestinal expression and function of Multidrug resistance-associated protein 2. Gastroenterology 2004;126:1044–53. [DOI] [PubMed] [Google Scholar]
- 23.Watkins BE, Esumi H, Wakabayashi K, et al. Fate and distribution of 2-amino-1-methyl-6-phenylimidazo[4,5- b]pyridine (PhIP) in rats. Carcinogenesis 1991;12:1073–8. [DOI] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–54. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–5. [DOI] [PubMed] [Google Scholar]
- 26.van Es HH, Goldhoorn BG, Paul-Abrahamse M, et al. Immunochemical analysis of uridine diphosphate-glucuronosyltransferase in four patients with the Crigler-Najjar syndrome type I. J Clin Invest 1990;85:1199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Geier A , Dietrich CG, Voigt S, et al. Effects of proinflammatory cytokines on rat organic anion transporters during toxic liver injury and cholestasis. Hepatology 2003;38:345–54. [DOI] [PubMed] [Google Scholar]
- 28.Morrow CS, Smitherman PK, Townsend AJ. Role of multidrug-resistance protein 2 in glutathione S-transferase P1- 1-mediated resistance to 4-nitroquinoline 1-oxide toxicities in HepG2 cells. Mol Carcinog 2000;29:170–8. [DOI] [PubMed] [Google Scholar]
- 29.Kaderlik KR, Minchin RF, Mulder GJ, et al. Metabolic activation pathway for the formation of DNA adducts of the carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in rat extrahepatic tissues. Carcinogenesis 1994;15:1703–9. [DOI] [PubMed] [Google Scholar]
- 30.Malfatti MA, Connors MS, Mauthe RJ, et al. The capability of rat colon tissue slices to metabolize the cooked-food carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine. Cancer Res 1996;56:2550–5. [PubMed] [Google Scholar]
- 31.de Waziers I , Cugnenc PH, Yang CS, et al. Cytochrome p 450 isoenzymes, epoxide hydrolase and glutathione transferases in rat and human hepatic and extrahepatic tissues. J Pharmacol Exp Ther 1990;253:387–94. [PubMed] [Google Scholar]
- 32.Koepke JP, Jones S, DiBona GF. Renal nerves mediate blunted natriuresis to atrial natriuretic peptide in cirrhotic rats. Am J Physiol 1987;252:R1019–23. [DOI] [PubMed] [Google Scholar]
- 33.Oswald M , Kullak-Ublick GA, Paumgartner G, et al. Expression of hepatic transporters OATP-C and MRP2 in primary sclerosing cholangitis. Liver 2001;21:247–53. [DOI] [PubMed] [Google Scholar]
- 34.Kullak-Ublick GA, Baretton GB, Oswald M, et al. Expression of the hepatocyte canalicular multidrug resistance protein (MRP2) in primary biliary cirrhosis. Hepatol Res 2002;23:78–82. [DOI] [PubMed] [Google Scholar]
- 35.Turesky RJ, Constable A, Fay LB, et al. Interspecies differences in metabolism of heterocyclic aromatic amines by rat and human P450 1A2. Cancer Lett 1999;143:109–12. [DOI] [PubMed] [Google Scholar]
- 36.Garner RC, Lightfoot TJ, Cupid BC, et al. Comparative biotransformation studies of MeIQx and PhIP in animal models and humans. Cancer Lett 1999;143:161–5. [DOI] [PubMed] [Google Scholar]
- 37.Lin DX, Lang NP, Kadlubar FF. Species differences in the biotransformation of the food-borne carcinogen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine by hepatic microsomes and cytosols from humans, rats, and mice. Drug Metab Dispos 1995;23:518–24. [PubMed] [Google Scholar]
- 38.Magagnotti C , Pastorelli R, Pozzi S, et al. Genetic polymorphisms and modulation of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP)-DNA adducts in human lymphocytes. Int J Cancer 2003;107:878–84. [DOI] [PubMed] [Google Scholar]