

Abstract
Background/Aim: Hepatic stellate cells (HSCs) express α-smooth muscle actin (αSMA) and acquire a profibrogenic phenotype upon activation by noxious stimuli. Insulin-like growth I (IGF-I) has been shown to stimulate HSCs proliferation in vitro, but it has been reported to reduce liver damage and fibrogenesis when given to cirrhotic rats.
Methods: The authors used transgenic mice (SMP8-IGF-I) expressing IGF-I under control of αSMA promoter to study the influence of IGF-I synthesised by activated HSCs on the recovery from liver injury.
Results: The transgene was expressed by HSCs from SMP8-IGF-I mice upon activation in culture and in the livers of these animals after CCl4 challenge. Twenty four hours after administration of CCl4 both transgenic and wild type mice showed similar extensive necrosis and increased levels of serum transaminases. However at 72 hours SMP8-IGF-I mice exhibited lower serum transaminases, reduced hepatic expression of αSMA, and improved liver morphology compared with wild type littermates. Remarkably, at this time all eight CCl4 treated wild type mice manifested histological signs of liver necrosis that was severe in six of them, while six out of eight transgenic animals had virtually no necrosis. In SMP8-IGF-I mice robust DNA synthesis occurred earlier than in wild type animals and this was associated with enhanced production of HGF and lower TGFβ1 mRNA expression in the SMP8-IGF-I group. Moreover, Colα1(I) mRNA abundance at 72 hours was reduced in SMP8-IGF-I mice compared with wild type controls.
Conclusions: Targeted overexpression of IGF-I by activated HSCs restricts their activation, attenuates fibrogenesis, and accelarates liver regeneration. These effects appear to be mediated in part by upregulation of HGF and downregulation of TGFβ1. The data indicate that IGF-I can modulate the cytokine response to liver injury facilitating regeneration and reducing fibrosis.
Keywords: carbon tetrachloride, HGF, smooth muscle actin, TGFβ, transgenic mice
Activation of hepatic stellate cells (HSCs), the main producers of liver collagen, involves induction of smooth muscle alpha actin (αSMA) and conversion from quiescent to proliferative, collagen expressing phenotype.1 A number of recent studies have focused attention on role of insulin-like growth factor I (IGF-I) in liver disease and HSC biology. In normal liver, IGF-I is synthesised at high levels in hepatocytes and in multiple non-parenchymal cell types2 including HSCs.3,4 Normal hepatocytes express 5–20 fold higher levels of IGF-I than non-parenchymal cells,2 and hepatocyte derived IGF-I is the major source of circulating IGF-I.5 Circulating IGF-I mediates growth of multiple tissues but appears to have little effect on growth of normal liver because normal hepatocytes express few IGF-I receptors.2,6 During chronic liver disease, serum IGF-I levels are decreased and provide a useful index of hepatocellular dysfunction and impaired nutritional status.1,3 Prior studies in a rat model of chronic, CCl4 induced liver cirrhosis indicate that systemic administration of IGF-I improves nutritional status and liver function7 and reduces oxidative damage and fibrosis.8
Hepatic stellate cells are known targets for IGF-I action. In cultured HSCs, IGF-I increases proliferation3,4,9–14 and collagen synthesis,8 providing indirect evidence that IGF-I could play a role in expansion of activated HSCs and in liver fibrosis. Quiescent HSCs, however, are continuously exposed to high local concentrations of IGF-I in normal liver and, yet, do not proliferate despite expression of high levels of type 1 IGF receptor (IGFIR).4,15 In addition, IGF-I stimulates production of hepatocyte growth factor (HGF) in hepatic stellate cells.16,17 HGF is not only a potent mitogen for hepatocytes18 but also appears to limit liver fibrosis in vivo.19 This suggests that IGF-I could indirectly act on HSC to stimulate the production of HGF which in turn could protect hepatocytes from injury or promote hepatocellular regeneration. Hence, the biological role of IGF-1 in the damaged liver has not yet been well defined and present concepts are mainly based on in vitro evidence.
It has been shown that HSCs can affect the rate of hepatocyte regeneration by regulating extracellular matrix (ECM) composition.1 Also, transforming growth factor beta 1 (TGFβ1), a major stimulus of ECM production in HSCs,1 has an inhibitory effect on replication of hepatocytes.20 Recent studies have demonstrated that interruption of TGFβ1 signalling improves liver fibrosis21,22 and stimulates liver regeneration,22 indicating that TGFβ1 strongly influences the biology of both HSCs and hepatocytes.
In this study we used transgenic mice (SMP8-IGF-I TG) in which the αSMA promoter drives the expression of rat IGF-I cDNA23 as a model to assess the role of HSC derived IGF-I on liver responses to acute CC14 induced injury in vivo. In this mouse, the SMP8-IGF transgene is not detectable in normal liver,23 presumably because αSMA positive cells are scarce. We hypothesised that activation of HSCs by CCl4 would induce expression of the SMP8-IGF-I transgene in the activated HSCs and would allow us to define the effects of HSC derived IGF-I on acute liver damage and repair in vivo.
METHODS
Animals
SMP8-IGF-I transgenic (TG) mice were derived as previously described.23 The SMP8-IGF-I TG mice have germline integration of a transgene composed of the murine αSMA promoter linked to a full length rat IGF-I cDNA. Hemizygous SMP8-IGF-I TG males, on the FVB-N genetic background, were bred with FVB-N WT females (Jackson Laboratories, West Grove, PA, USA) to derive TG and WT mice for analyses. Study protocols were in full compliance with NIH guidelines for humane animal care.
CCl4 treatment
Ether sedated, 50–60 day old, sex matched TG and WT littermates were given either a single dose of CCl4 (Sigma, St Louis, MO, USA) or the same dose of corn oil by gavage (3.5 ml/kg of body weight in a 1:1 mixture with corn oil).24 In an initial study, mice were killed at 72 hours after CCl4, a time when previous studies have established full activation of HSCs in the mouse.25 In a follow up experiment, mice were killed at 3, 8, 12, 24, 48, or 72 hours after CCl4 or vehicle treatment. In this study, bromodeoxyuridine (BrdU; Sigma) was given as a single intraperitoneal injection (120 mg/kg) at 90 minutes before sacrifice. Mice were anaesthetised with Pentobarbital (Veterinary Laboratories Inc, Lanefa, KS, USA) and killed by exsanguination.
IGF-I radioimmunoassay
Plasma IGF-I concentration was measured by radioimmunoassay after removal of IGF binding proteins by C18 cartridge chromatography (Sep-Pak; Waters, Milford, MA, USA) as previously described.26
Liver enzymes
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured using standard clinical automatic analyser (Hitachi 736).
RNA extraction and analyses
Liver RNA was extracted by a guanidine thiocyanate homogenisation, cesium chloride sedimentation method as detailed in Lund et al.27 20 μg of total RNA was analysed by northern blot hybridisation using a 32P-labelled (Amersham, Arlington Heights, IL, USA) antisense RNA probe complementary to rat IGF-I28 and a transgene specific RNA probe complementary to the SV40 3′ untranslated region present within the SMP8-IGF-I transgene product.23 A mouse PL7 cRNA, encoding a ribosomal protein, was used as a control for RNA loading.
Protein isolation and western blot
Frozen liver samples were homogenised in 10 mM Tris buffer (pH 7.4) containing a cocktail of protease inhibitors (Sigma). HSC were lysed in 10 mM Hepes buffer (pH 7.9) also containing a protease inhibitor cocktail. Protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA). Extracts corresponding to 50 μg of protein were separated on 12% non-denaturing SDS polyacrylamide gels and transferred to PVDF membranes (Immobilon-P; Millipore, Bedford, MA, USA), as previously described.29 Filters were blocked in 10% non-fat dry milk in TBS-T (20 mM Tris-HCl pH 7.6, 137 mM NaCl and 1% Tween) before incubation with primary antibodies in 5% milk in TBS-T, for 60 minutes at room temperature. After washing, membranes were incubated with HRP conjugated secondary antibodies (Jackson Immuno Research Laboratories, West Grove, PA, USA), and developed using enhanced chemiluminescence light (ECL) detection kit (Amersham Biosciences, Piscataway, NY, USA). Primary antibodies used at 1:100 dilution were mouse monoclonal antibody to αSMA (Clone A4; Dako, Carpinteria, CA, USA), and to BCL-XL (Santa Cruz Biotechnology, Santa Cruz, CA, USA), and polyclonal antibodies to HGF (R&D Systems, Minneapolis, MN, USA) and IGF-I (Santa Cruz Biotechnology). A policlonal antibody against tubulin (Santa Cruz Biotechnology) was used as a protein loading control.
Immunohistochemistry
Formalin fixed, paraffin embedded liver blocks were sectioned at 6 μm and stained with haematoxylin and eosin to evaluate morphology or with Masson’s trichrome and Sirius red to detect collagen deposition. Immunohistochemistry to localise αSMA and BrdU was performed as previously described.29
Scoring of liver necrosis
Liver necrosis was graded on coded haematoxylin and eosin stained sections by an experienced pathologist blinded to treatment groups. Scoring was as follows: grade O, no visible necrosis; grade I, focal necrosis of less than 25% of the liver lobule area; grade II, confluent centrilobular necrosis of 25–50%; grade III, necrosis of 50–75%; grade IV, necrosis of more than 75% of the liver lobule30
Isolation and culture of HSCs
HSCs from untreated TG and WT mice were isolated and purified as described previously.25 HSCs were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal serum, 100 IU/ml penicillin, and 100 μg/ml streptomycin. HSCs were plated out at a density of 106 cells/cm2. Quiescent (newly isolated) and activated (day 14 in culture) HSCs were studied. Total RNA from cultured HSCs was prepared using TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) according to the manufacturer’s instructions.
RT-PCR
Expression of the transgene in cultured HSC was measured using standard RT-PCR techniques. 1 μg of liver or HSC derived total RNA was reverse transcribed to complementary DNA (cDNA) using oligo(dT) and avian myeloblastosis virus reverse transcriptase (Promega, Madison, WI, USA). The sequence of a sense primer corresponding to αSMA 5′ UT and an antisense primer corresponding to SV40 3′ UT of the SMP8-IGF-I transgene, as well as specific primers used to detect β-actin, are listed in table 1 ▶. Amplification reactions were carried out as previously described31 using the GeneAmp PCR System 2400 (Perkin-Elmer, Norwalk, CT, USA).
Table 1.
Oligonucleotide sequences used in RT-PCR reactions
| mRNA | Oligomers | |
| SMP8-IGF-I | Sense | 5′-GTG GCT TCT AGT GCT GAC G-3′ |
| Antisense | 5′-CCA CTC AGG CGT ATC AGT G-3′ | |
| TGFβ1 | Sense | 5′-GAG TGG TTT GTT TGA GAT GT-3′ |
| Antisense | 5′-GGT TCG TGC ATC CAT TTC CA-3′ | |
| Colα1(I) | Sense | 5′-GCA AAG AGT AGT CTA CAT GTC TAG-3′ |
| Antisense | 5′-CCT ACA TCT TCT GAG TTT GG-3′ | |
| Fibronectin | Sense | 5′-TGT GAC AAC TGC CGT AGA CC-3′ |
| Antisense | 5′-GAC CAA CTG TCA CCA TTG AGG-3′ | |
| β-actin | Sense | 5′-GTG ACG AGG CCC AGA GCA AGA G-3′ |
| Antisense | 5′-AGG GGC CGG ACT CAT CGT-3′ | |
| 28s | Sense | 5′-AGA AGG GCA AAA GCT CGC TT-3′ |
| Antisense | 5′-AGC AGG ATT ACC ATG GCA AC-3′ | |
Real-time RT-PCR was also performed to quantify expression of the mRNAs of procollagen α1(I) (Colα 1(I)), fibronectin, transforming growth factor beta 1 (TGFβ1), and the 28 ribosomal subunit (28s) using specific, intron spanning, primers to avoid co-amplification of genomic DNA (table 1 ▶). For real-time quantitative RT-PCR, reverse transcription of total liver RNA to cDNA was done at 42°C for one hour using random primers (Boehringer Ingelheim, Germany) and RTase (Gibco, Grand Island, NY, USA). During PCR amplification, the cDNA was incubated with a mix containing SYBR Green I, a dye specific for double stranded DNA (LightCycler-FastStart DNA Master SYBR Green I; Roche Diagnostics, Barcelona, Spain). Amplification conditions using a thermocycler (LightCycler; Roche Molecular Biochemicals) were one step denaturation for 10 minutes at 95°C, followed by multiple cycles of denaturation for 15 seconds at 95°C, annealing for five seconds at 61°C, and extension for 13 seconds at 72°C. The number of cycles were: 26, 39, and 44 for 28s, Colα1(I), and TGFβ1, respectively. The specification of each PCR product was established based on its melting temperatures (LightCycler software version 3.39; Roche Molecular Biochemicals, Indianapolis, IN, USA), and appropriate size was verified on agarose gels. To prepare standards, PCR generated fragments were isolated from agarose gels, purified (GeneClean II; Q-Biogene, Carlsbad, CA, USA), cloned into the pGEM T-Easy vector (Promega, Madison, WI, USA) and sequenced (ABI PRISM 310 Genetic Analyzer; Perkin Elmer, USA). Standard curves were generated from serial dilutions of control linearised plasmids to obtain relative quantitation. Results are presented as ratios of the relative concentrations of Colα1(I) and TGFβ1 to the 28s values.
Statistical analyses
Two way ANOVA was used to test for significance of effects of the transgene (WT v TG) and CC14 treatment, and interactions between transgene and treatment which indicate an effect of transgene on reponses to CC14. Post hoc, pairwise comparisons were performed using Duncan’s test. Mann-Whitney U test for non-parametric inference was used when appropriate, as indicated in the text.
RESULTS
Induction of SMP8-IGF-I mRNA
In the liver of TG mice the expression of SMP8-IGF-I transgene, as assessed by RT-PCR, occurred as soon as eight hours after CCl4 administration and tended to increase along the 72 hours of the study period (fig 1A ▶). Northern blot analysis of liver samples obtained from WT and TG mice 72 hours after vehicle or CC14 administration showed that the induction of the transgene occurred only in TG mice that received CC14 (fig 1B ▶). To confirm that the transgene is specifically expressed in activated HSCs, we examined transgene expression by RT-PCR in HSCs isolated from WT and TG mice. HSCs were examined in quiescence and after 14 days of culture on plastic to induce activation.25,32 We observed that SMP8-IGF-I mRNA was detected only in activated TG HSCs (fig 1C ▶).
Figure 1.
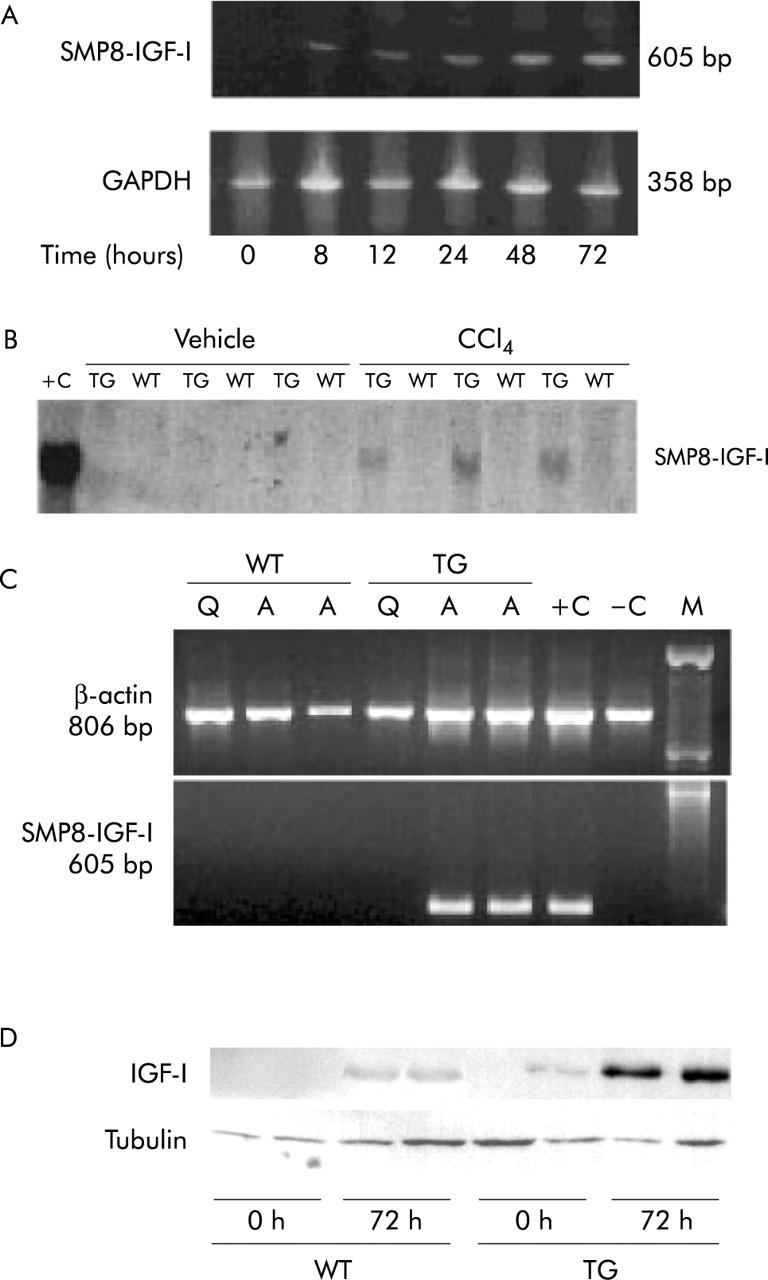
(A) Time course of liver expression of SMP8-IGF-I transgene in TG mice after CCl4 treatment, measured by RT-PCR. (B) Autoradiogram of a northern blot of liver RNA probed with a transgene specific probe. Total RNA was extracted from the livers of six WT and six TG mice at 72 hours after administration of vehicle or CCl4, as described in Methods. 20 μg of total RNA from small intestine (a site of strong transgene expression in smooth muscle cells) of TG mice was used as a positive control (+C). (C) RT-PCR products obtained with oligomers specific for the SMP8-IGF-I transgene and control β-actin. RT-PCR was performed on RNA isolated from quiescent (Q) or activated (A) HSCs. HSCs were activated by culture during 14 days on a plastic surface. HSCs isolated from three WT and three TG mice were pooled for extraction of RNA used for RT-PCR. +C, RT-PCR product from total RNA from small intestine of SMP8-IGF-I TG mouse; −C, negative control; M, markers. (D) Representative western blot of IGF-I in liver tissue of WT and TG mice at 0 h and 72 h after CCl4. Tubulin expression was used as loading protein control.
Assays of plasma IGF-I revealed a significant (p<0.01) decline in circulating IGF-I concentrations in both WT and TG mice at 24 hours after CC14, but levels returned to normal by 72 hours (table 2 ▶). At this time point, western blot for IGF-I protein in the injured liver showed that the levels of this growth factor was more intense in TG mice than in WT animals (fig 1D ▶), indicating that under conditions of liver damage the hepatic production of IGF-I is higher in the former group of animals.
Table 2.
Plasma IGF-I concentration at 24 and 72 hours after intragastric administration of CCl4
| Vehicle | 24 hours | 72 hours | |
| WT | 417.7 (13.7) | 226.6 (57.8)* | 408.3 (90.1) |
| SMP8-IGF-I TG | 422.3 (7.8) | 227.0 (48.1)* | 426.4 (51.2) |
Data represent the mean (SEM) obtained from three mice in each group. Two way ANOVA revealed significant effect of CCl4 treatment on the concentrations of circulating IGF-I at 24 hours (p<0.01), but no effect of genotype. *Significantly different from vehicle treated mice.
Liver function and morphology
The livers of WT or TG mice receiving vehicle were histologically unremarkable. At 24 and 48 hours after CCl4 administration the typical centrilobular hepatocellular necrosis induced by this compound could be seen in both groups of animals (fig 2 ▶).33,34 However, at 72 hours the liver lesion was largely resolved in TG mice while significant necrosis could still be observed in WT animals (fig 2 ▶). Thus, at this time point six out of eight WT mice showed severe hepatic necrosis (grade III) and the other two had moderate necrosis (grade II) while among TG mice six out of eight had virtually no necrosis and other two exhibited necrosis of moderate degree (grade II).
Figure 2.
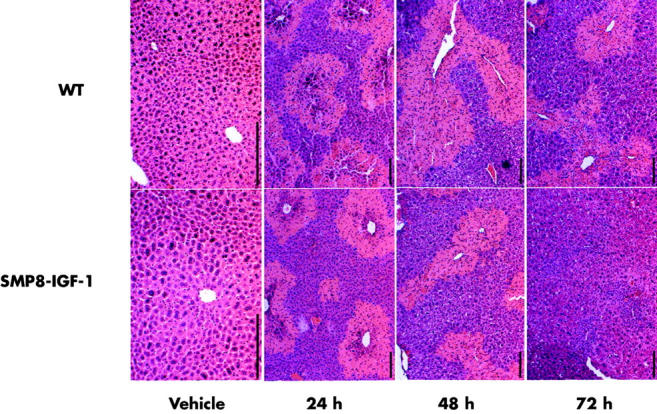
Liver morphology in WT and SMP8-IGF-I TG mice treated with vehicle and after CCl4 treatment. Livers were harvested at 24, 48, or 72 hours after intragastric administration of vehicle or CCl4. Portions of the liver were fixed, sectioned, and stained with haematoxylin and eosin. Representative photomicrographs from eight mice per group are shown. Scale bar = 200 μm.
In agreement with these histological findings, serum biochemistry demonstrated a dramatic increase in transaminase levels (>1000 IU/l) at 24 hours after CCl4 ingestion in the two groups of animals, but recovery from hepatic damage was more rapid in TG mice, which showed at 48 and 72 hours values of serum transaminases significantly lower than those found in WT littermates (table 3 ▶).
Table 3.
Serum levels of AST and ALT in wild type and SMP8-IGF-I mice after CCl4 administration
| 0 hours | 24 hours | 48 hours | 72 hours | |
| AST | ||||
| WT | 20 (2) | 32580 (1957) | 8087 (487) | 2255 (366) |
| SMP8-IGF-I TG | 21 (6) | 24020 (4951) | 2507 (158)* | 1167 (479)* |
| ALT | 0 hours | 24 hours | 48 hours | 72 hours |
| WT | 29 (8) | 54600 (1693) | 7623 (1051) | 1880 (253) |
| SMP8-IGF-I TG | 33 (7) | 42100 (3864) | 4780 (501)* | 520 (178) |
Values are expressed as IU/l and represent the mean (SEM) obtained from three to six mice in each group. *p<0.05 compared with the respective group of WT mice.
Liver regeneration
In order to label proliferating cells, BrdU was administered 90 minutes before sacrifice in animals examined at 24, 48, and 72 hours after CCl4 treatment (fig 3 ▶). In both WT and TG animals, BrdU positive cells were very rarely encountered at 24 hours after injury. At 48 hours we observed an intense regeneration wave in TG mice which showed a frequency of BrdU labelled hepatocytes significantly higher than the WT littermates (218 (31) v 137 (7) positive nuclei per high power field, p<0.05). At 72 hours, when the healing was almost complete, the number of BrdU positive nuclei started declining in TG mice (103 (23)), while it still was on the rise in WT animals (182 (14)). These data indicate the occurrence of an accelerated hepatocyte regeneration in TG mice, which takes place with an anticipation of about 24 hours with respect WT littermates.
Figure 3.

Immunohistochemical staining of the liver of WT and TG mice following CCl4 treatment. Ninety minutes before sacrifice, at the times indicated in the figure, mice received intraperitoneal injection of BrdU to visualise proliferating cells. Livers were harvested, processed, and stained for BrdU as described in Methods. Representative photomicrographs are shown; arrows show positive nuclei. Scale bar = 200 μm.
The transgenic IGF-I could stimulate liver regeneration either directly or through other factors involved in the regulation of hepatocyte proliferation. Mature HGF protein, a complete mitogen for hepatocytes,18 can be detected in the liver as early as six hours after CCl435 and reaches its maximal values at 24 hours.4,14 Using western blot analysis of livers at 24 hours after CCl4 administration we found that the hepatic levels of HGF were higher in TG mice than in WT animals (fig 4A ▶ and B). The levels of HGF in hepatic tissue still remained higher in TG than in WT mice at 72 hours after the toxic challenge (data not shown). In order to determine whether the increased production of HGF might be derived from enhanced production of this factor by activated transgenic HSC, we analysed by western blot the production of HGF in extracts of transgenic and wild type HSC in a quiescent status or after activation by culturing during 14 days. As shown in figure 4C ▶, we found that HGF was produced only by activated TG HSC, but not by WT HSC, nor by quiescent HSC. These data show that transgenic IGF-I may act autocrine/paracrine fashion to activate HGF production in HSC.
Figure 4.
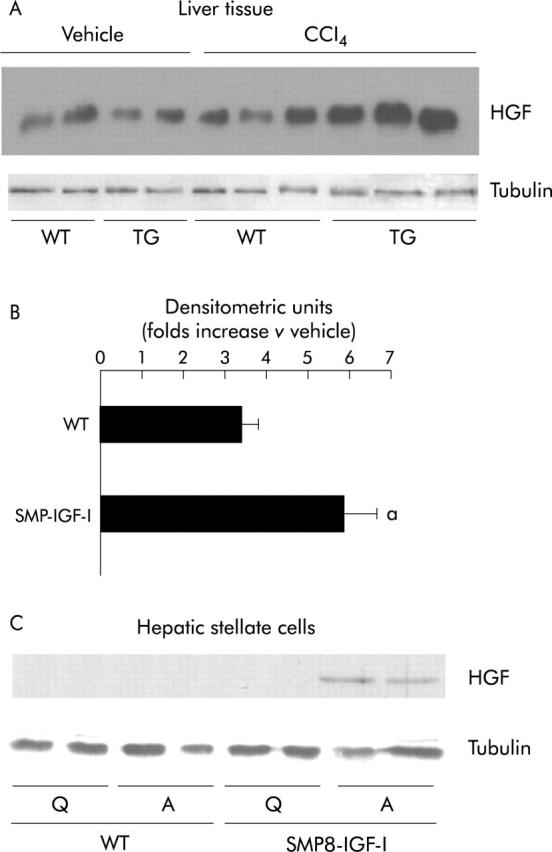
(A) Western blot analysis of HGF levels in the livers of CCl4 treated WT and SMP8-IGF-I TG mice. Proteins were extracted, as described in Methods, from livers harvested at 24 hours following intragastric administration of vehicle or CCl4. Western blot was performed using anti-HGF polyclonal antibody, and tubulin was used as protein loading control. (B) The intensity of each band was quantified using an image analyser. Results are expressed as the ratio between the density signal in CCl4 treated mice to the density signal in vehicle treated mice set arbitrarily at 1.0. Data represent the mean (SEM) obtained from three mice per group. a: significantly different (p<0.05) from WT mice (Mann-Whitney U test). (C) Representative photomicrograph of western blot analyses of HGF levels in HSC obtained from WT and SMP8-IGF-I transgenic mice, and activated by culturing during 14 days; tubulin was used as protein loading control. Q: Quiescent HSC; A: Activated HSC.
High levels of TGFβ1 inhibit liver regeneration20 whereas low levels have been shown to have a permissive effect on hepatocyte replication.36,37 In our study we found that compared with WT, TG animals tended to express lower levels of TGFβ1 mRNA during the whole study period, the differences being statistically significant (p<0.05, Mann-Whitney U-test) at 24 hour after CCl4 administration (fig 5A ▶). At this time point TGFβ1 mRNA expression was increased (relative to animals that received vehicle) 5.84 (0.72) fold in WT and only 2.04 (0.47) fold in TG. TGFβ1 mRNA expression was also analysed in HSC from TG and WT mice activated by culture on plastic for 14 days. We found that HSC obtained from TG mice showed a three fold reduction in TGFβ1 mRNA expression compared with those obtained from WT mice (fig 5B ▶). Again these observations indicate that the transgenic IGF-I can repress the synthesis of the profibrogenic cytokine TGFβ1 by HSC either directly or indirectly through stimulation of HGF production.
Figure 5.

(A) Real-time RT-PCR analyses of TGFβ1 mRNA values in the livers of WT and SMP8-IGF-I TG mice. Livers were harvested at indicated times following treatment with vehicle or CCl4. Total RNA was extracted and processed as detailed in Methods. Following amplification, mRNA levels were standardised to those of the housekeeping gene 28s. Results are expressed as the ratio between relative values of TGFβ1 mRNA in CCl4 treated mice to those of TGFβ1 mRNA in vehicle treated mice set arbitrarily at 1.0. Data represent the mean (SEM) from three to six mice per group analysed at each time point. b: significantly different (p<0.01) from vehicle treated group; c: significantly different (p<0.05) from TG mice. (B) Real time RT-PCR analyses of TGFβ1 mRNA levels in HSC obtained from WT and transgenic mice, activated during 14 days in culture. Total RNA was extracted and processed as detailed in Methods. Units were calculated from the formula: 2ΔCt×10000 (ΔCt = number of cycles of housekeeping gene minus number of cycles of TGFβ1). Data represent the mean (SEM) from 10–12 experiments. Statistical significances are shown in the figure. (C) Real-time quantitative RT-PCR analyses of expression of procollagen α1(I) (left panel) and fibronectin (right panel) in the livers of WT and SMP8-IGF-I TG mice harvested at indicated time points following vehicle or CCl4 treatment (see methodological details in (A)). Data represent the mean (SEM) from three to six mice per group analysed at each time point. a: significantly different (p<0.05) from vehicle-treated group; b: significantly different (p<0.05) from TG mice (Mann-Whitney U test).
Collagen and fibronectin expression
Acute CCl4 induced liver injury is accompanied by transient elevation in the expression of Colα1(I) mRNA.25,38 In our study Colα1(I) mRNA levels were significantly (p<0.05) increased in both WT and TG mice at 48 and 72 h after CCl4 treatment (fig 5C ▶, left). At 72 hours, the increase in Colα1(I) mRNA values (relative to mice receiving vehicle) was significantly lower (p<0.05) in CCl4 treated TG mice than in CCl4 treated WT littermates (fig 5C ▶, left). Despite upregulation of Colα1(I) mRNA, no collagen accumulation in the liver in either genotype was visualised using Masson’s trichrome or Sirius red stains in our model of acute CCl4 liver injury.
The expression of fibronectin, another ECM component, remained largely unchanged except at 72 hours, when its mRNA levels (relative to mice given vehicle) were significantly lower (p<0.05) in CCl4 treated TG animals than in WT mice that received the hepatotoxin (fig 5C ▶, right).
Activation of HSC
αSMA staining was negligible in the liver of WT or TG mice that received vehicle. By contrast a dramatic increase in the number of αSMA positive cells was present in the livers of WT mice given CCl4. Interestingly, fewer αSMA positive cells were detected in TG mice at 72 hours after CCl4 ingestion and were located around the central vein (fig 6 ▶, top). To confirm the immunohistochemical findings, αSMA expression in the liver extracts was analysed by immunoblot. As illustrated in figure 6 ▶ (bottom), αSMA was not detectable in vehicle treated WT or TG mice whereas it was strongly induced after CCl4 administration in WT (seven out of seven) but in only one out of six TG mice examined.
Figure 6.

Top: Immunohistochemical staining of the liver with antibody specific for αSMA. Sections of the livers from WT and SMP8-IGF-I TG mice, harvested at 72 hours following vehicle or CCl4 treatment, were stained with anti-αSMA monoclonal antibody as described in Methods. Representative photomicrographs from eight mice per group are shown. Scale bar = 200 μm. Bottom: Representative western blot of analyses of αSMA (46 kDa) levels in the livers obtained from the same experiment. Tubulin western blot is included as a loading protein control.
Apoptosis of activated HSCs has been reported in some models during resolution of liver injury.39,40 We performed TUNEL assays to determine if increased apoptosis accounted for the lower numbers of activated HSC in TG mice. No TUNEL positive HSCs were found in livers of vehicle or CCl4 treated WT or TG mice (data not shown) indicating that apoptosis of HSC is minimal in the mouse CC14 model of acute liver injury. Furthermore, we evaluated by western blot the expression of the antiapoptotic protein BCL-XL, whose levels are usually decreased in liver damage.41 We found that the reduction induced by CCl4 at 72 hours after toxic ingestion in both genotypes was less intense in TG than in WT mice (fig 7 ▶).
Figure 7.

Representative western blot of BCL-XL levels in the livers obtained from WT and SMP8-IGF-I TG mice, harvested at 72 hours following vehicle or CCl4 treatment. Tubulin western blot is included as a loading protein control.
DISCUSSION
In the present study transgene expression was not detectable in the normal liver of SMP8-IGF-I TG mice, despite the fact that αSMA is present in the scant smooth muscle cells of the intrahepatic branches of the hepatic artery. A low level of transgenic expression in these structures which represent a minimal part of the hepatic mass might determine that the abundance of SMP8-IGF-I mRNA in normal transgenic liver is below the detection threshold of the PCR technique used in our study. By contrast we show that in SMP8-IGF-I TG mice the transgene is induced in the liver after CCl4 injury and in isolated HSCs upon activation, thus demonstrating that the αSMA promoter is a useful tool to deliver expression of selected genes specifically to activated HSCs. This model allowed us to analyse the effect of HSC derived IGF-I on the liver response to injury.
Massive necrosis of hepatocytes after CCl4 ingestion is followed by a replicative response which is orchestrated by a highly complex cytokine cascade. The whole reparative process is usually completed within five days.42 Our results show that injury induced expression of the SMP8-IGF-I gene in activated HSCs in vivo was not sufficient to clearly diminish initial damage by CCl4, but did lead to accelerated liver regeneration, improved liver morphology and biochemistry, and fewer activated HSCs by 72 hours after CCl4, indicating an effect of the transgene in the improvement of the recovery after injury. This rapid cure seems to result from stimulation of liver cell regeneration, but we cannot completely exclude the contribution of a direct or indirect effect of IGF-I on the modulation of the intensity of hepatocellular damage.
We have observed that IGF-I protein can be detected by western blot at higher levels in the liver of TG mice than in WT mice. As the concentration of circulating IGF-I is similar in WT and TG mice after injury, it seems that improved healing observed in TG mice may result from the autocrine/paracrine effects of transgene derived IGF-I induced in HSCs upon activation.
SMP8 IGF-I-derived IGF-I could act indirectly by modulating the synthesis of other growth factors which regulate hepatocellular proliferation. We focused our attention on HGF, a complete mitogen for hepatocytes that increases transiently in the injured liver,18,43 and TGFβ1, a known inhibitor of hepatocyte growth which is induced after CCl4 injury.20 In our study we observed, at 24 hours after injury, a more intense increase of HGF in the liver of TG mice than in WT animals. The increase in HGF expression persisted, although at lower levels at 72 hours. This finding suggests that the HSC derived IGF-I may accelerate healing at least in part by enhancing the HGF response to injury. This interpretation is supported by earlier findings demonstrating induction of HGF by IGF-I in HSCs in vitro16,17 and by our data showing that HSC from TG mice produce more HGF upon activation than wild type HSC.
TGFβ1 is a multifunctional cytokine which on one hand activates HSCs and promotes fibrogenesis1 and on the other hand inhibits hepatocyte proliferation.20 The observed decrease in TGFβ1 in TG mice (fig 5 ▶) could contribute to accelerated liver regeneration36,37 as well as to the reduction in the number of αSMA positive HSCs.21,25 This reduction does not appear to be due to increased apoptosis of HSC because no apoptotic HSC were detected either in WT or TG mice, and BCL-XL was more intensely expressed in TG than in WT livers (fig 7 ▶). The process of activation of HSCs involves a TGFβ1 independent initiation phase followed by TGFβ1 dependent perpetuation phase.25 The reduced number of αSMA positive HSCs found in TG mice could reflect attenuated perpetuation, consistent with the low TGFβ1 mRNA levels observed both in liver tissue and HSC in these animals. The exact mechanism of inhibition of TGFβ1 mRNA expression by IGF-I remains unknown, but could involve the ability of IGF-I to increase antioxidant activity of catalase in the liver44 or lower numbers of activated HSCs expressing TGFβ.
Our results show that IGF-I induction in HSCs attenuates collagen expression during acute liver injury in TG mice. It seems likely that the decrease in collagen mRNA may result from different factors including reduced injury, enhanced production of HGF (a hepatotrophic factor known to suppress hepatic fibrogenesis19) and decreased TGFβ1 mRNA. In spite of increased collagen gene expression, neither WT nor TG mice showed signs of accumulation of collagen in this model of acute liver injury (data not shown) which is consistent with reports in other strains.24
Although we were able to detect IGF-I by western blot in livers from TG mice after toxic challenge, we could not definitively localise SMP8-IGF-I transgene expression solely to HSCs. Attempts to localise transgene by in situ hybridisation or histochemistry proved problematic because of high non-specific binding of cRNA probes or IGF-I antibodies to necrotic tissue and because the transgene specific probe is small and has limited sensitivity. We cannot exclude that the transgene expression is induced in other cell types as well as in HSCs, such as vascular smooth muscle cells. In any case, the dramatic improvement in repair in SMP8-IGF-I TG, despite similar circulating IGF-I as in WT, indicates that the effect reflects local transgene induction in HSCs and possibly other cell types with resulting largely paracrine activities. It is also important to note that the beneficial effect of HSC derived IGF-I during acute injury does not anticipate what would be the role of HSC derived IGF-I in fibrosis associated with chronic injury. We have attempted to compare the responses of SMP8-IGF-I TG and WT to chronic CCl4 induced injury and fibrosis but this FVB strain appears resistant to fibrosis (unpublished data).
In conclusion, our study demonstrates that the SMP8 promoter is a useful tool to deliver genes of interest to activated HSCs in vitro and to injured liver and, likely, to activated HSCs in vivo. SMP8-IGF-I induction in the liver in vivo during CCl4 injury accelerates liver regeneration and this is associated with increased production of HGF, reduced expression of TGFβ1, and reduced collagen mRNA expression. Together these data support a beneficial role of HSC derived IGF-I in tissue repair after acute liver injury.
Acknowledgments
This work was facilitated by the Center for Gastrointestinal Biology and Disease (NIH grant DK-34987) and supported by NIH grants DK-02402 (to JBP), DK-47769 (to PKL), Chinese Government Award (to SL), a travel grant from Echébano Foundation (to SS), and grants from Instituto Salud Carlos III C03/02, Fondo Investigaciones Sanitarias 01/0843 (to CMR-O), and Ministerio Ciencia y Tecnologia SAF 2002–0327 (to JP).
Abbreviations
28s, 28 ribosomal subunit
αSMA, smooth muscle α actin
ALT, alanine aminotransferase
AST, aspartate aminotransferase
BrdU, bromodeoxyuridine
CCl4, carbon tetrachloride
cDNA, complementary DNA
Colα1(I), procollagen α1(I)
ECM, extracellular matrix
HGF, hepatocyte growth factor
HSCs, hepatic stellate cells
IGF-I, insulin-like growth factor I
IGFIR, IGF-I receptor 1
mRNA, messenger RNA
RT-PCR, reverse transcription polymerase chain reaction
TG, transgenic mice
TGFβ1, transforming growth factor beta 1,
UT, untranslated region
WT, wild type mice
SS and SL contributed equally to this work.
REFERENCES
- 1.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000;275:2247–50. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann EM, Li L, Hoyt EC, et al. Cell-specific localization of insulin-like growth factor binding protein mRNAs in rat liver. Am J Physiol 2000;278:G447–G457. [DOI] [PubMed] [Google Scholar]
- 3.Pinzani M , Abboud HE, Aron DC. Secretion of insulin-like growth factor-I and binding proteins by rat liver fat-storing cells: regulatory role of platelet-derived growth factor. Endocrinology 1990;127:2343–9. [DOI] [PubMed] [Google Scholar]
- 4.Scharf JG, Knittel T, Dombrowski F, et al. Characterization of the IGF axis components in isolated rat hepatic stellate cells. Hepatology 1998;27:1275–84. [DOI] [PubMed] [Google Scholar]
- 5.Yakar S , Liu J-L, Stannard B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A 1999;96:7324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caro JF, Poulos J, Ittoop O, et al. Insulin-like growth factor I binding in hepatocytes from human liver, human hepatoma, and normal, regenerating, and fetal rat liver. J Clin Invest 1988;81:976–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Picardi A , Deoliveira AC, Muguerza B, et al. Low doses of insulin-like growth factor-I improve nitrogen retention and food efficiency in rats with early cirrhosis. J Hepatol 1997;26:191–202. [DOI] [PubMed] [Google Scholar]
- 8.Svegliati-Baroni G , Ridolfi F, Di Sario A, et al. Insulin and insulin-like growth factor-1 stimulate proliferation and type I collagen accumulation by human hepatic stellate cells: differential effects on signal transduction pathways. Hepatology 1999;29:1743–51. [DOI] [PubMed] [Google Scholar]
- 9.Bachem MG, Meyer D, Schafer W, et al. The response of rat liver perisinusoidal lipocytes to polypeptide growth regulator changes with their transdifferentiation into myofibroblast-like cells in culture. J Hepatol 1993;18:40–52. [DOI] [PubMed] [Google Scholar]
- 10.Gentilini A , Feliers D, Pinzani M, et al. Characterization and regulation of insulin-like growth factor binding proteins in human hepatic stellate cells. J Cell Physiol 1998;174:240–50. [DOI] [PubMed] [Google Scholar]
- 11.Gentilini A , Marra F, Gentilini P, et al. Phosphatidylinositol-3 kinase and extracellular signal-regulated kinase mediate the chemotactic and mitogenic effects of insulin-like growth factor-I in human hepatic stellate cells. J Hepatol 2000;32:227–34. [DOI] [PubMed] [Google Scholar]
- 12.Gressner AM, Brenzel A, Vossmeyer T. Hepatocyte-conditioned medium potentiates insulin-like growth factor (IGF) 1 and 2 stimulated DNA synthesis of cultured fat storing cells. Liver 1993;13:86–94. [DOI] [PubMed] [Google Scholar]
- 13.Gressner AM, Lahme B, Brenzel A. Molecular dissection of the mitogenic effect of hepatocytes on cultured hepatic stellate cells. Hepatology 1995;22:1507–18. [PubMed] [Google Scholar]
- 14.Zindy F , Lamas E, Schmidt S, et al. Expression of insulin-like growth factor II (IGF-II) and IGF-II, IGF-I and insulin receptors mRNAs in isolated non-parenchymal rat liver cells. J Hepatol 1992;14:30–4. [DOI] [PubMed] [Google Scholar]
- 15.Brenzel A , Gressner AM. Characterization of insulin-like growth factor (IGF)-I-receptor binding sites during in vitro transformation of rat hepatic stellate cells to myofibroblasts. Eur J Clin Chem Clin Biochem 1996;34:401–9. [DOI] [PubMed] [Google Scholar]
- 16.Skrtic S , Wallenius K, Gressner AM, et al. Insulin-like growth factor signaling pathways in rat hepatic stellate cells: importance for deoxyribonucleic acid synthesis and hepatocyte growth factor production. Endocrinology 1999;140:5729–35. [DOI] [PubMed] [Google Scholar]
- 17.Skrtic S , Wallenius K, Ekberg S, et al. Insulin-like growth factors stimulate expression of hepatocyte growth factor but not transforming growth factor beta1 in cultured hepatic stellate cells. Endocrinology 1997;138:4683–9. [DOI] [PubMed] [Google Scholar]
- 18.Burr AW, Toole K, Chapman C, et al. Anti-hepatocyte growth factor antibody inhibits hepatocyte proliferation during liver regeneration. J Pathol 1998;185:298–302. [DOI] [PubMed] [Google Scholar]
- 19.Matsuda Y , Matsumoto K, Yamada A, et al. Preventive and therapeutic effects in rats of hepatocyte growth factor infusion on liver fibrosis/cirrhosis. Hepatology 1997;26:81–9. [DOI] [PubMed] [Google Scholar]
- 20.Russell WE, Coffey RJJ, Ouellette AJ, et al. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci U S A 1988;85:5126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.George J , Roulot D, Koteliansky VE, et al. In vivo inhibition of rat stellate cell activation by soluble transforming growth factor beta type II receptor: a potential new therapy for hepatic fibrosis. Proc Natl Acad Sci U S A 1999;96:12719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ueno H , Sakamoto T, Nakamura T, et al. A soluble transforming growth factor beta receptor expressed in muscle prevents liver fibrogenesis and dysfunction in rats. Hum Gene Ther 2000;11:33–42. [DOI] [PubMed] [Google Scholar]
- 23.Wang J , Niu W, Nikiforov Y, et al. Targeted overexpression of IGF-I evokes distinct patterns of organ remodeling in smooth muscle cell tissue beds of transgenic mice. J Clin Invest 1997;100:1425–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Z , Wakil AE, Rockey DC. Strain-specific differences in mouse hepatic wound healing are mediated by divergent T helper cytokine responses. Proc Natl Acad Sci U S A 1997;94:10663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hellerbrand C , Stefanovic B, Giordano F, et al. The role of TGFß1 in initiating hepatic stellate cell activation in vivo. J Hepatol 1999;30:77–87. [DOI] [PubMed] [Google Scholar]
- 26.Davenport ML, D’Ercole AJ, Underwood LE. Effect of maternal fasting on fetal growth, serum insulin-like growth factors (IGFs), and tissue IGF messenger ribonucleic acids. Endocrinology 1990;126:2062–7. [DOI] [PubMed] [Google Scholar]
- 27.Lund PK, Moats-Staats BM, Hynes MA, et al. Somatomedin-C/insulin-like growth factor-I and insulin-like growth factor-II mRNAs in rat fetal and adult tissues. J Biol Chem 1986;261:14539–44. [PubMed] [Google Scholar]
- 28.Thissen JP, Pucilowska JB, Underwood LE. Differential regulation of insulin-like growth factor I (IGF-I) and IGF binding protein-1 messenger ribonucleic acids by amino acid availability and growth hormone in rat hepatocyte primary culture. Endocrinology 1994;134:1570–6. [DOI] [PubMed] [Google Scholar]
- 29.Pucilowska JB, McNaughton KK, Mohapatra NK, et al. IGF-I and procollagen α1(I) are coexpressed in a subset of mesenchymal cells in active Crohn’s disease. Am J Physiol 2000;279:G1307–22. [DOI] [PubMed] [Google Scholar]
- 30.Zhang LP, Takahara T, Yata Y, et al. Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: role of stellate cells. J Hepatol 1999;31:703–11. [DOI] [PubMed] [Google Scholar]
- 31.Simmons JG, Pucilowska JB, Lund PK. Autocrine and paracrine actions of intestinal fibroblast-derived insulin-like growth factors. Am J Physiol 1999;276:G817–27. [DOI] [PubMed] [Google Scholar]
- 32.Rockey DC, Boyles JK, Gabbiani G, et al. Rat hepatic lipocytes express smooth muscle actin upon activation in vivo and in culture. J Submicrosc Cytol Pathol 1992;24:193–203. [PubMed] [Google Scholar]
- 33.Cameron GR, Karunarante WAE. Carbon tetrachloride cirrhosis in relation to liver regeneration. J Pathol Bacteriol 1936;62:1–20. [Google Scholar]
- 34.Smuckler EA. Structural and functional changes in acute liver injury. Environ Health Perspect 1976;15:13–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shimizu M , Hara A, Okuno M, et al. Mechanism of retarded liver regeneration in plasminogen activator—deficient mice: impaired activation of hepatocyte growth factor after Fas-mediated massive hepatic apoptosis. Hepatology 2001;33:569–76. [DOI] [PubMed] [Google Scholar]
- 36.Date M , Matsuzaki K, Matsushita M, et al. Modulation of transforming growth factor beta function in hepatocytes and hepatic stellate cells in rat liver injury. Gut 2000;46:719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura T , Sakata R, Ueno T, et al. Inhibition of transforming growth factor beta prevents progression of liver fibrosis and enhances hepatocyte regeneration in dimethylnitrosamine-treated rats. Hepatology 2000;32:247–55. [DOI] [PubMed] [Google Scholar]
- 38.Maher JJ, McGuire RF. Extracellular matrix gene expression increases preferentially in rat lipocytes and sinusoidal endothelial cells during hepatic fibrosis in vivo. J Clin Invest 1990;86:1641–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iredale JP, Benyon RC, Pickering J, et al. Mechanisms of spontaneous resolution of rat liver fibrosis. Hepatic stellate cell apoptosis and reduced hepatic expression of metalloproteinase inhibitors. J Clin Invest 1998;102:538–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saile B , Knittel T, Matthes N, et al. CD95/CD95L-mediated apoptosis of the hepatic stellate cell. A mechanism terminating uncontrolled hepatic stellate cell proliferation during hepatic tissue repair. Am J Pathol 1997;151:1265–72. [PMC free article] [PubMed] [Google Scholar]
- 41.Kovalovich K , Li W, DeAngelis R, et al. Interleukin-6 protects against Fas-mediated death by establishing a critical level of anti-apoptotic hepatic proteins FLIP, Bcl-2, and Bcl-xL. J Biol Chem 2001;276:26605–13. [DOI] [PubMed] [Google Scholar]
- 42.Yamada Y , Fausto N. Deficient liver regeneration after carbon tetrachloride injury in mice lacking type 1 but not type 2 tumor necrosis factor receptor. Am J Pathol 1998;152:1577–89. [PMC free article] [PubMed] [Google Scholar]
- 43.Maher JJ. Cell-specific expression of hepatocyte growth factor in liver. Upregulation in sinusoidal endothelial cells after carbon tetrachloride. J Clin Invest 1993;91:2244–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castilla-Cortázar I , García M, Muguerza B, et al. Hepatoprotective effects of insulin-like growth factor I in rats with carbon tetrachloride-induced cirrhosis. Gastroenterology 1997;113:1682–91. [DOI] [PubMed] [Google Scholar]