

Rather than a unique disorder, Crohn’s disease is regarded as a common denominator for various immune mediated inflammatory diseases of the gastrointestinal tract. These various subforms of Crohn’s disease are characterised by T cell mediated tissue destruction in the gut. In contrast with autoimmune diseases where autoantibodies react with self antigens, inflammation in Crohn’s disease is directed against antigens that are mainly derived from the environment.1 Although it may sound trivial, this adjustment in the understanding of the pathogenetic mechanisms has a vast number of consequences for the search of the disease causing factors and identification of therapeutic targets.
CROHN’S PHENOTYPES
The various subforms of Crohn’s disease have not been recognised until recently. Historically, colonic Crohn’s disease was not distinguished from ulcerative colitis. Approximately 30 years ago, the first studies highlighted the diverse natural history of colonic and ileal Crohn’s disease.2 Inherent differences in disease behaviour (obstructing versus perforating) were later recognised by a group of physicians at Mount Sinai Hospital in New York.3 Biological discrimination between various subgroups of Crohn’s patients led to a proposal for a phenotypic classification,4 which was taken up and further developed and evaluated by an international working group, leading to the Vienna classification.5 Accordingly, Crohn’s disease is separated into three categories: age at diagnosis, location, and behaviour (table 1 ▶). Several teams have now tested the Vienna classification in their databases6–8 and found that it is feasible and reproducible. The behaviour and age categories earned some criticism which will be discussed later. The location category was found to be very useful and was also stable over time, with only one of seven patients having changes over a 10 year observation period.7
Table 1.
Vienna classification of Crohn’s disease
| Age at diagnosis |
| Age when diagnosis of Crohn’s disease was first definitively established by radiology, endoscopy, pathology, or surgery. |
| • A1—<40 y |
| • A2—⩾40 y |
| Location |
| Maximum extent of disease involvement at any time before the first resection. Minimum involvement for a location is defined as any aphthous lesion or ulceration. Mucosal erythema and oedema are insufficient. For classification, at least both a small bowel and a large bowel examination are required. |
| • L1—Terminal ileum |
| - Disease limited to the terminal ileum (the lower third of the small bowel) with or without spill over into the caecum. |
| • L2—Colon |
| - Any colonic location between the caecum and rectum with no small bowel or upper gastrointestinal (GI) involvement. |
| • L3—Ileocolon |
| - Disease of the terminal ileum with or without spill over into the caecum and any location between the ascending colon and rectum. |
| • L4—Upper GI |
| - Any disease location proximal to the terminal ileum (excluding the mouth) regardless of additional involvement of the terminal ileum or colon. |
| Behaviour |
| • B1—Non-stricturing non-penetrating |
| - Inflammatory disease which never has been complicated at any time in the course of disease. |
| • B2—Stricturing: |
| - Stricturing disease is defined as the occurrence of constant luminal narrowing demonstrated by radiological, endoscopic, or surgical-pathological methods, with prestenotic dilatation or obstructive signs/symptoms without the presence of penetrating disease at any time in the course of disease. |
| • B3—Penetrating: |
| - Penetrating disease is defined as the occurrence of intra-abdominal or perianal fistulas, inflammatory masses, and/or abscesses at any time in the course of disease. Perianal ulcers are also included. Excluded are postoperative intra-abdominal complications and perianal skin tags. |
Adapted from Gasche and colleagues.5
CLASSIFICATION BY AETIOLOGY
The best classification would be by cause of disease, but unfortunately this is not known. However, there is overwhelming indirect evidence for the presence of environmental triggers, although the specific antigen(s) that drives the immune reaction in Crohn’s disease is still obscure. Cigarette smoking is the only confirmed external factor involved in the pathogenesis of Crohn’s disease. It has been linked to disease progression and unfavourable disease outcome across the world. The underlying mechanism is incompletely understood.9 Smoking seems to be a disease aggravating rather than a disease causing factor. In fact, smokers are more likely to develop strictures or fistulae.10 No other external trigger has been generally recognised.
CROHN’S GENOTYPES
Apart from the disease causing environment, variations in the human genome contribute to disease susceptibility and shape the clinical phenotype. The higher frequency of Crohn’s disease in certain ethnic groups (particularly in Caucasians), familial aggregation, and a greater disease concordance in monozygotic twins are strong indicators of the impact of genetic factors in disease pathogenesis. However, the genetic trait of Crohn’s disease is complex, with genetic heterogeneity and incomplete phenotype penetrance. To date, seven susceptibility loci have been identified cosegregating preferentially with disease phenotype11 but no dominant inflammatory bowel disease gene has been detected. Genetic variations are loading the gun but the environmental factor still pulls the trigger.
This paradigm was further supported by identification of caspase activation and recruitment domain 15 (CARD15) variants that confer susceptibility to Crohn’s disease.12 What have we learned about the function of CARD15?
CARD15/NOD2
In 2001, Nod2 was identified as a Nod1/Apaf-1 homologous gene which mapped to chromosome 16q12.13 Later the gene was renamed CARD15 by the Human Genome Organisation (HUGO) nomenclature committee.12,14,15 CARD15 consists of 11 constant exons and an alternative in the 5′ region. Its 1040 amino acids are structured into four distinct domains: two N terminal CARDs, a central nucleotide binding domain (NBD), and a C terminal leucine rich repeat domain (LRR). CARDs are responsible for interaction with CARDs from other proteins, such as RIP-like interacting CLARP kinase (RICK), and potentially with proteins of the apoptosis pathway. NBD is responsible for self oligomerisation. The LRR domain is required for peptidoglycan (PGN) induced nuclear factor κB (NFκB) activation.
Three variants (SNP8 [R702W], SNP12 [G908R], and SNP13 [L1007fs]) within the LRR were initially described to be associated with Crohn’s disease.12 Screening of the entire coding region of CARD15 identified another 30 non-conservative missense mutations.16 The three common mutations are present in up to 50% of Crohn’s patients but also in up to 20% of healthy individuals, and they account for 82% of mutated alleles.16 CARD15 variants seem to be rather disease specific because they do not confer risk to other chronic inflammatory or autoimmune diseases.17,18 Variants in the NBD were found to be associated with Blau syndrome, a rare autosomal dominant disorder characterised by early onset granulomatous arthritis, uveitis, and skin rash with camptodactyly.17
One important observation is that CARD15 mutations are thought to have a dose dependent effect as mutated homozygotes and compound heterozygotes are found more frequently in Crohn’s patients than expected. The three common CARD15 mutations are neither necessary nor sufficient for expression of the disease phenotype. In Caucasians, they do not explain more than 20% of the genetic predisposition to Crohn’s disease. This also varies between different European countries as a high diversity exists in different European populations (table 2 ▶). The absence of CARD15 variants in Asian and sub-Saharan African populations precludes any role for CARD15 variants in Crohn’s disease in these populations.19
Table 2.
Diversity of caspase activation and recruitment domain 15 (CARD15) allele frequencies in Europe
| Population | Controls/Crohn’s disease (%) | ||
| SNP8 (R702W) | SNP12 (G908R) | SNP13 (L1007fs) | |
| Finland47 | 1.8/3.3 | 0.0/0.6 | 1.7/4.8 |
| Norway48 | 2.8/3.5 | 1.2/0.9 | 1.2/2.6 |
| Scotland34 | –/8.0 | –/2.8 | –/4.2 |
| Ireland49 | 4.0/7.0 | 1.0/3.0 | 1.0/4.0 |
| Great Britain33 | 5.2/12.5 | 1.4/3.3 | 1.6/9.4 |
| Germany50 | –/– | –/– | 3.3/23.7 |
| Germany48 | 4.8/9.9 | 0.7/4.0 | 4.1/13.7 |
| Poland51 | –/– | –/– | 3.6/– |
| Netherlands52 | –/– | 3.0/4.3 | 1.0/8.5 |
| Spain53 | 2.2/7.1 | 1.1/4.2 | 2.3/7.4 |
| Italy54 | –/– | –/– | 1.3/11.7 |
| Italy55 | 5.9/9.0 | 1.4/4.3 | 2.3/6.3 |
| Crete56 | –/– | –/– | 1.5/2.7 |
| European mix16 | 4.0/11.0 | 1.0/6.0 | 1.9/11.0 |
| European mix57 | 4.2/11.4 | 0.2/4.2 | 3.4/14.6 |
SNP, single nucleotide polymorphism; –, not investigated.
The CARD15 pathway
CARD15 is one of the mammalian bacteria sensing proteins that is part of innate immunity leading to activation of the NFκB pathway (fig 1 ▶). In the intestine, CARD15 is preferentially expressed in Paneth cells and macrophages. Because of the overwhelming presence of bacteria in proximity to the mucosal lining, toll-like receptor function is downregulated in intestinal epithelial cells thereby preventing undesired permanent immune activation.20 Therefore, prevention of bacterial invasion in the gut relies mainly on CARD15 and Nod1. CARD15 and Nod1 have an overlapping recognition pattern for bacterial PGNs, with CARD15 being broader than Nod1. Thus CARD15 has a distinct protective role in the intestinal tract because loss of function mutations are only partially compensated for by Nod1.21,22
Figure 1.

Caspase activation and recruitment domain 15 (CARD15) is mainly expressed in the granulocyte-macrophage lineage (including dendritic cells) and also in Paneth cells of the intestinal tract.13,25,36,37 CARD15 is activated by binding of naturally occurring muropeptides, enzymatic degradation products of peptidoglycans (PGNs) derived from bacterial cells walls.22 Their presence triggers CARD15 oligomerisation (as indicated by three overlapping CARD15 molecules) and recruitment of RIP-like interacting CLARP kinase (RICK) via CARD-CARD interaction. RICK then activates the nuclear factor κB inhibitor (IκB) kinase complex (IKK) via phosphorylation of IKKγ. The ΙΚΚ complex next phosphorylates IκB resulting in nuclear factor κB (NFκB) translocation to the nucleus and transcriptional activation of NFκB responsive genes such as proinflammatory cytokines or defensins.60
Apart from activation of NFκB, Nod1 and CARD15 are capable of activating caspase-1 and caspase-9.23,24 However, the mechanism responsible for excess activity of NFκB in Crohn’s lesions is not fully understood and may theoretically be related to insufficient induction of apoptosis by CARD15 mutants. In return, the CARD15 promoter contains NFκB response elements mediating its transcriptional activation after tumour necrosis factor α (TNF-α) stimulation.25,26 TNF-α dependent expression of CARD15 can be enhanced but not independently induced by interferon γ (IFN-γ). In contrast, Nod1 responds to IFN-γ27 but not to TNF-α. This positive feedback loop of CARD15 was found in infected intestinal epithelial cells and may explain increased CARD15 expression in epithelial cells surrounding colonic Crohn’s lesions.28,29
GENOTYPE-PHENOTYPE CORRELATIONS IN CROHN’S DISEASE
Several groups have independently studied the association between CARD15 variants and Crohn’s disease phenotype (summarised in table 3 ▶). Appreciable differences were detected in double mutation carriers. They showed unanimously that CARD15 variants only confer susceptibility to ileal (L1 and L3 in the Vienna classification) but not colonic (L2 in the Vienna classification) Crohn’s disease.30,31 Also, CARD15 polymorphisms are not associated with ulcerative colitis or indeterminate colitis, two distinct colonic forms of inflammatory bowel disease.12,31,32 Carriers are further characterised by a younger age at onset (A1 in the Vienna classification).16,33 This fits with a general observation that the relative importance of genetic (inherited) and environmental (acquired) factors is shifted towards genes in younger patients and towards environment in the elderly. Some studies found a relation between CARD15 and complicating (stricturing or perforating) disease. Multivariate analyses however identified smoking habits and anti-saccharomyces antibody profiles as the underlying factor.10,16,34,35 Intestinal biopsies from double mutant carriers are reported to display more granulomas,16 underlining the link between CARD15 and granuloma formation, as also observed in Blau syndrome.
Table 3.
Genotype-phenotype correlations in Crohn’s disease
| L1 (ileal) | L2 (colonic) | L3 (ileocolonic) | |
| Cuthberth32 | + | – | + |
| Lesage16 | + | – | – |
| Murillo52 | +/− | – | +/− |
| Ahmad33 | + | – | + |
| Vermeire58 | + | – | + |
| Hampe48 | + | +/− | + |
| Abreu59 | + | – | + |
| Mendoza53 | + | – | – |
| Helio47 | + | – | – |
| Bairead49 | + | – | – |
| Giachino55 | + | – | – |
+/−, trend but not statistically significant.
The relationship between CARD15 mutations and a certain Crohn’s disease phenotype may serve as proof of principle: Crohn’s disease is not a single disease but a common denominator for various immune mediated inflammatory diseases of the gastrointestinal tract. We may ask how CARD15 mutations can determine the ileal location of Crohn’s disease. The reason for this may be related to the predominant expression of CARD15 in Paneth cells.36,37 Paneth cells, on the other hand, are most numerous in the terminal ileum and are critically important in enteric antibacterial defence (fig 2 ▶). These findings suggest a role for CARD15 in the regulation of the Paneth cell mediated response against intestinal bacteria and a plausible mechanism explaining the selective association of CARD15 mutations with ileal Crohn’s disease. The impaired capacity of Crohn’s disease associated CARD15 mutations to sense the intracellular presence of bacterial cell wall components (that is, PGN) may result in overgrowth and invasion of certain gut microbes, perhaps through impaired production of human defensins leading to chronic inflammation.38
Figure 2.
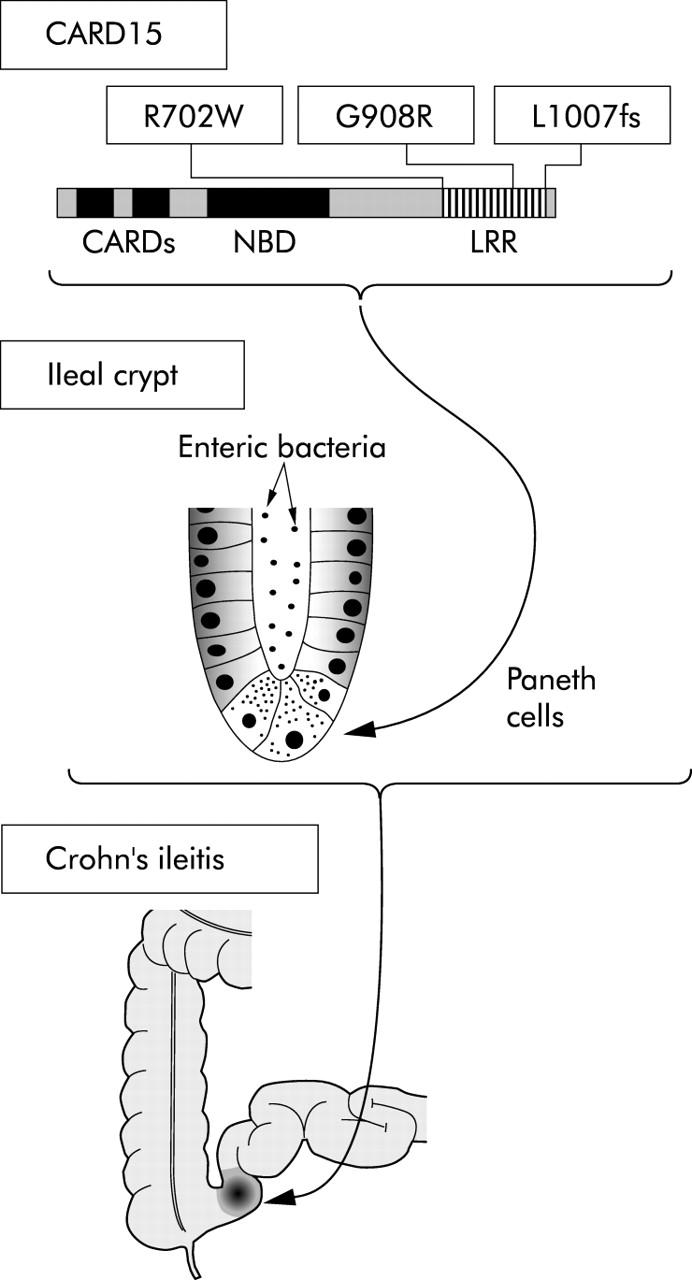
Three caspase activation and recruitment domain 15 (CARD15) variants (R702W, G908R, and L1007fs) within the leucine rich repeat (LRR) were initially described to genetically predispose to the development of Crohn’s disease.12 These variants are disease specific because they do not confer risk to other chronic inflammatory or autoimmune diseases.17,18 In the intestinal tract, CARD15 is primarily expressed in Paneth cells, which are critical in enteric antibacterial defence.28 In CARD15 mutant individuals, nuclear factor κB (NFκB) activation is reduced and thus the necessary antibacterial response, such as expression of defensins, does not occur.61,62 Chronic NFκB activation through alternative pathways is regarded as secondary to the lack of an appropriate antibacterial response.21 Paneth cells are most numerous in the terminal ileum and seem to be of local importance. A CARD15 deficit in Paneth cells, as present in homozygous or compound heterozygous mutants, is phenotypically related to Crohn’s ileitis. NBD, nucleotide binding domain.
DOES PHENOTYPING AND GENOTYPING HAVE RELEVANCE IN CLINICAL PRACTICE?
Assessment of disease phenotype
There is little controversy that the phenotypic discrimination of Crohn’s subgroups is a key to the clinical management of patients. Information regarding the patient’s age and sex, disease location, and disease progression has been collected long before the Vienna classification was established because it is needed for appropriate patient care. For example, information on disease location is crucial for prescribing topical therapy39; information on disease progression (mild versus aggressive) is pertinent when considering immunosuppressive therapy. The Vienna classification did not introduce new disease variables but rather provided a structured format to collect a minimal amount of information on the most important variables.
The Vienna classification has however drawn legitimate criticism for some shortcomings. Notable among these is the observation that the behavioural classification progresses with disease duration.7,8 Definitions for the aggressive behaviour phenotype need to be adjusted in the future.40 Suggestions include implementing a certain point in time after diagnosis (for example, five or 10 years) to finally establish a complicating (B2 or B3) disease phenotype. When setting this point at five years post diagnosis, one could expect approximately 48% of cases to be B1, 12% B2, and 40% B3, and at 10 years approximately 30% B1, 14% B2, and 66% B3.8 Another suggestion relates to the separation of perianal and intra-abdominal fistulising disease which are currently grouped within the B3 category.41 Thereby, perianal disease supersedes any internal disease characteristic and forces a case to be classified as B3. Perforating disease behaviour was primarily recognised as an indicator of aggressive disease that requires repeated operations.3 This feature needs further attention, particularly in relation to perianal disease which is often the most debilitating aspect of Crohn’s disease.42 However, the behaviour category aims to depict the degree, speed, and quality of tissue destruction, a clinically obvious and relevant biological distinction between individual patients. The age category has been criticised for its arbitrary cut off at age 40 years. It is difficult however to identify solid arguments for other cut off points. The higher proportion of patients with colonic disease (L2) within the A2 group is a good indicator that this separation has biological meaning.5 For now, the Vienna classification remains a simple tool to minimally describe patient populations in clinical trials and eases communication between physicians in clinical practice.
Assessment of disease genotype
Although laboratory assays for detection of CARD15 variants are trivial and robust, its widespread application in clinical practice should be strongly reconsidered because of jurisdictional, ethical, and economical limitations. The central question is: What is the benefit for the individual who undergoes genetic testing?
Identifying individuals at risk
One of the advantages of genetic testing in general, is the possibility of identifying individuals at risk of developing a certain disease, thereby facilitating appropriate action for disease prevention. With regard to Crohn’s disease, we have to question the general risk of developing this phenotype in the first instance? Such risk depends on the prevalence of Crohn’s disease in the population studied. In most Western European or North American countries the incidence now plateaus at 6–7/100 000/year, with a disease prevalence of approximately 150–180/100 000.43 Canada, exceptionally and without any obvious explanation, has a significantly higher incidence of 15/100 000/year and a prevalence of 200/100 000.44 In accordance, the lifetime risk for development of Crohn’s disease was estimated to be between 0.15% and 0.3%.
What is the risk of developing Crohn’s disease in individuals with a disease associated CARD15 variant? Compared with individuals carrying two wild-type alleles, the odds ratio of disease development was estimated to be 2–3 for heterozygous carriers and 20–40 for homozygotes or compound heterozygotes.12,14,15,32 Therefore, the lifetime risk for heterozygote individuals increases to 0.45–0.9%, and for homozygote or compound heterozygote individuals to 3.0–12.0%. However, even in the highest incidence areas (such as Canada) and using the highest risk estimates (3 and 40), only 1 of 111 heterozygous and 1 of 8 homozygous or compound heterozygous individuals will develop the disease phenotype. In addition, in the average Caucasian population (assuming a combined allele frequency (sum of the three major CARD15 SNPs) of approximately 7%), one has to screen 200 individuals to identify a single homozygous or compound heterozygous individual. Genetic variations on different genes may add some risk to phenotype penetrance. Most likely however the clue to this remains undiscovered within the environment of Western countries. Because of these shortcomings, screening for CARD15 variants in Western (and also any other) populations is currently not useful.
Is there a role for genetic testing in Crohn’s family members? Genetic testing in gastroenterology has become a precious tool in several familial diseases, including hereditary cancer syndromes such as familial adenomatous polyposis and hereditary non-polyposis colorectal cancer. On the one hand, genetic testing allows for exclusion of transmission of a certain mutation and thereby relieves the individual at risk from the burden of early cancer development. On the other hand, preventive actions such as drug therapy, surveillance endoscopy, or proctocolectomy can be offered to mutation carriers. As CARD15 variants cover only 20% of genetic susceptibility to Crohn’s disease and because of the low phenotype penetrance, it seems currently unreasonable to perform genetic testing in Crohn’s families. In other words, neither a negative (double wild-type) nor a positive test result (double mutant) carries a certainty in predicting phenotype penetrance. Most importantly, no measures are available to prevent Crohn’s disease in individuals at risk. All that physicians can do is recommend that the patient not smoke. This however does not require genetic testing.
Differential diagnosis
Can CARD15 variants help to confirm or exclude the diagnosis of Crohn’s disease, specifically in the case of indeterminate colitis? Again, negative tests do not rule out a diagnosis of Crohn’s disease and a positive test does not prove its presence. Moreover, the ulcerative colitis-like Crohn’s phenotype (L2 in the Vienna classification) is very uncommon in patients carrying two mutations.31 This low frequency of CARD15 mutations in Crohn’s colitis suggests that CARD15 genotyping has a limited role in this situation.
Pharmacogenetics
Genetic variations are considered important variables which may define a response or adverse event of drug therapy. Our wish is to tailor drug therapy to the genetic background of individual patients. For example, some centres perform thiopurine methyltransferase genotyping for improving 6-mercaptopurine and azathioprine safety in Crohn’s disease. So far, CARD15 variants were studied in relation to infliximab therapy45,46 without detecting any association with treatment response or side effects. Drug development programmes intend to interfere with the specific genetic defect in Crohn’s disease but no drug has been marketed yet that interacts with the CARD15 pathway.
Genotyping within clinical trials
The fact that Crohn’s disease is not a single entity is readily appreciated when studying genetic susceptibility. CARD15 double mutants are a distinct genetically defined Crohn’s subgroup. As clinical trials generally report on the composition of the study cohort, future trials could implement the frequency of CARD15 mutations. This could improve transferring the results of a certain study into other patient groups. A different response of CARD15 carriers to certain therapies may be observed in the future and will then allow stratification of patients on the basis of genotyping. Apart from the use of the Vienna classification, we therefore endorse subgroup analysis on the basis of CARD15 genetics in clinical trials
FINAL REMARKS
Identification of CARD15 variants as inheritable susceptibility factors in Crohn’s disease defines a milestone in the understanding of disease pathogenesis. CARD15 has provided a logical explanation for some of the phenomenon observed within families or twins. It has opened a door towards unravelling the network of Crohn’s pathogenesis. So far we have learned that it relates to ileal disease location, possibly through its selective expression in Paneth cells and regulation of antibacterial host defence. The pathogenetic understanding of CARD15 functions may open new avenues to specific preventive measures and therapies that interfere with the underlying disease mechanism and that are tailored to the patient’s genotype. Until then, physicians have to rely on clinical phenotype and their proper judgement of the patient’s symptoms.
Supplementary Material
Acknowledgments
CG is supported by grants P15314 from the Austrian Science Fund (FWF) and OeNB10543 from the Austrian National Bank Anniversary Fund for the Promotion of Scientific Research and Teaching.
Conflict of interest: None declared.
REFERENCES
- 1.Mayer L . Comment from the editors. Gastroenterology 2003;125:1574. [DOI] [PubMed] [Google Scholar]
- 2.Farmer RG, Hawk WA, Turnbull RB Jr. Clinical patterns in Crohn’s disease: a statistical study of 615 cases. Gastroenterology 1975;68:627–35. [PubMed] [Google Scholar]
- 3.Greenstein AJ, Lachman P, Sachar DB, et al. Perforating and non-perforating indications for repeated operations in Crohn’s disease: evidence for two clinical forms. Gut 1988;29:588–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sachar DB, Andrews HA, Farmer RG, et al. Proposed classification of patient subgroups in Crohn’s disease. Gastroenterol Int 1992;5:141–54. [Google Scholar]
- 5.Gasche C , Scholmerich J, Brynskov J, et al. A simple classification of Crohn’s disease: report of the Working Party for the World Congresses of Gastroenterology, Vienna 1998. Inflamm Bowel Dis 2000;6:8–15. [DOI] [PubMed] [Google Scholar]
- 6.Freeman HJ. Application of the Vienna classification for Crohn’s disease to a single clinician database of 877 patients. Can J Gastroenterol 2001;15:89–93. [DOI] [PubMed] [Google Scholar]
- 7.Louis E , Collard A, Oger AF, et al. Behaviour of Crohn’s disease according to the Vienna classification: changing pattern over the course of the disease. Gut 2001;49:777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cosnes J , Cattan S, Blain A, et al. Long-term evolution of disease behavior of Crohn’s disease. Inflamm Bowel Dis 2002;8:244–50. [DOI] [PubMed] [Google Scholar]
- 9.Cosnes J , Carbonnel F, Beaugerie L, et al. Effects of cigarette smoking on the long-term course of Crohn’s disease. Gastroenterology 1996;110:424–31. [DOI] [PubMed] [Google Scholar]
- 10.Louis E , Michel V, Hugot JP, et al. Early development of stricturing or penetrating pattern in Crohn’s disease is influenced by disease location, number of flares, and smoking but not by NOD2/CARD15 genotype. Gut 2003;52:552–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duerr RH. Update on the genetics of inflammatory bowel disease. J Clin Gastroenterol 2003;37:358–67. [DOI] [PubMed] [Google Scholar]
- 12.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001;411:599–603. [DOI] [PubMed] [Google Scholar]
- 13.Ogura Y , Inohara N, Benito A, et al. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J Biol Chem 2001;276:4812–18. [DOI] [PubMed] [Google Scholar]
- 14.Hampe J , Cuthbert A, Croucher PJ, et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 2001;357:1925–8. [DOI] [PubMed] [Google Scholar]
- 15.Ogura Y , Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001;411:603–6. [DOI] [PubMed] [Google Scholar]
- 16.Lesage S , Zouali H, Cezard JP, et al. CARD15/NOD2 mutational analysis and genotype-phenotype correlation in 612 patients with inflammatory bowel disease. Am J Hum Genet 2002;70:845–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miceli-Richard C , Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001;29:19–20. [DOI] [PubMed] [Google Scholar]
- 18.Miceli-Richard C , Zouali H, Lesage S, et al. CARD15/NOD2 analyses in spondylarthropathy. Arthritis Rheum 2002;46:1405–6. [DOI] [PubMed] [Google Scholar]
- 19.Nemeth M , Grundtner P, Willheim C, et al. The pattern of evolution and worldwide distribution of CARD15 variants as evidence for local selection pressure in the historic European populations. Gastroenterology 2004;126 (suppl 2) :A351. [Google Scholar]
- 20.Philpott DJ, Girardin SE, Sansonetti PJ. Innate immune responses of epithelial cells following infection with bacterial pathogens. Curr Opin Immunol 2001;13:410–16. [DOI] [PubMed] [Google Scholar]
- 21.Inohara N , Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem 2003;278:5509–12. [DOI] [PubMed] [Google Scholar]
- 22.Girardin SE, Boneca IG, Viala J, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003;278:8869–72. [DOI] [PubMed] [Google Scholar]
- 23.Bertin J , Nir WJ, Fischer CM, et al. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-kappa B. J Biol Chem 1999;274:12955–8. [DOI] [PubMed] [Google Scholar]
- 24.Inohara N , Koseki T, del Peso L, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappa B. J Biol Chem 1999;274:14560–7. [DOI] [PubMed] [Google Scholar]
- 25.Gutierrez O , Pipaon C, Inohara N, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J Biol Chem 2002;277:41701–5. [DOI] [PubMed] [Google Scholar]
- 26.Rosenstiel P , Fantini M, Brautigam K, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology 2003;124:1001–9. [DOI] [PubMed] [Google Scholar]
- 27.Hisamatsu T , Suzuki M, Podolsky DK. Interferon-gamma augments CARD4/NOD1 gene and protein expression through interferon regulatory factor-1 in intestinal epithelial cells. J Biol Chem 2003;278:32962–8. [DOI] [PubMed] [Google Scholar]
- 28.Hisamatsu T , Suzuki M, Reinecker HC, et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 2003;124:993–1000. [DOI] [PubMed] [Google Scholar]
- 29.Berrebi D , Maudinas R, Hugot JP, et al. Card15 gene overexpression in mononuclear and epithelial cells of the inflamed Crohn’s disease colon. Gut 2003;52:840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gasche C , Alizadeh BZ, Pena AS. Genotype-phenotype correlations: how many disorders constitute inflammatory bowel disease? Eur J Gastroenterol Hepatol 2003;15:599–606. [DOI] [PubMed] [Google Scholar]
- 31.Hugot JP, Zouali H, Lesage S. Lessons to be learned from the NOD2 gene in Crohn’s disease. Eur J Gastroenterol Hepatol 2003;15:593–7. [DOI] [PubMed] [Google Scholar]
- 32.Cuthbert AP, Fisher SA, Mirza MM, et al. The contribution of NOD2 gene mutations to the risk and site of disease in inflammatory bowel disease. Gastroenterology 2002;122:867–74. [DOI] [PubMed] [Google Scholar]
- 33.Ahmad T , Armuzzi A, Bunce M, et al. The molecular classification of the clinical manifestations of Crohn’s disease. Gastroenterology 2002;122:854–66. [DOI] [PubMed] [Google Scholar]
- 34.Walker LJ, Aldhous MC, Drummond HE, et al. Anti-Saccharomyces cerevisiae antibodies (ASCA) in Crohn’s disease are associated with disease severity but not NOD2/CARD15 mutations. Clin Exp Immunol 2004;135:490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mow WS, Vasiliauskas EA, Lin YC, et al. Association of antibody responses to microbial antigens and complications of small bowel Crohn’s disease. Gastroenterology 2004;126:414–24. [DOI] [PubMed] [Google Scholar]
- 36.Lala S , Ogura Y, Osborne C, et al. Crohn’s disease and the NOD2 gene: a role for Paneth cells. Gastroenterology 2003;125:47–57. [DOI] [PubMed] [Google Scholar]
- 37.Ogura Y , Lala S, Xin W, et al. Expression of NOD2 in Paneth cells: a possible link to Crohn’s ileitis. Gut 2003;52:1591–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wehkamp J , Harder J, Weichenthal M, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal α-defensin expression. Gut 2004;53:1639–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rutgeerts P , Lofberg R, Malchow H, et al. A comparison of budesonide with prednisolone for active Crohn’s disease. N Engl J Med 1994;331:842–5. [DOI] [PubMed] [Google Scholar]
- 40.Sachar DB. Behaviour of Crohn’s disease according to the Vienna classification. Gut 2002;51:614–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Brant SR, Picco MF, Achkar JP, et al. Defining complex contributions of NOD2/CARD15 gene mutations, age at onset, and tobacco use on Crohn’s disease phenotypes. Inflamm Bowel Dis 2003;9:281–9. [DOI] [PubMed] [Google Scholar]
- 42.Present DH. Crohn’s fistula: current concepts in management. Gastroenterology 2003;124:1629–35. [DOI] [PubMed] [Google Scholar]
- 43.Loftus EV Jr, Silverstein MD, Sandborn WJ, et al. Crohn’s disease in Olmsted County, Minnesota, 1940–1993: incidence, prevalence, and survival. Gastroenterology 1998;114:1161–8. [DOI] [PubMed] [Google Scholar]
- 44.Bernstein CN, Blanchard JF, Rawsthorne P, et al. Epidemiology of Crohn’s disease and ulcerative colitis in a central Canadian province: a population-based study. Am J Epidemiol 1999;149:916–24. [DOI] [PubMed] [Google Scholar]
- 45.Vermeire S , Louis E, Rutgeerts P, et al. NOD2/CARD15 does not influence response to infliximab in Crohn’s disease. Gastroenterology 2002;123:106–11. [DOI] [PubMed] [Google Scholar]
- 46.Mascheretti S , Hampe J, Croucher PJ, et al. Response to infliximab treatment in Crohn’s disease is not associated with mutations in the CARD15 (NOD2) gene: an analysis in 534 patients from two multicenter, prospective GCP-level trials. Pharmacogenetics 2002;12:509–15. [DOI] [PubMed] [Google Scholar]
- 47.Helio T , Halme L, Lappalainen M, et al. CARD15/NOD2 gene variants are associated with familially occurring and complicated forms of Crohn’s disease. Gut 2003;52:558–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hampe J , Grebe J, Nikolaus S, et al. Association of NOD2 (CARD 15) genotype with clinical course of Crohn’s disease: a cohort study. Lancet 2002;359:1661–5. [DOI] [PubMed] [Google Scholar]
- 49.Bairead E , Harmon DL, Curtis AM, et al. Association of NOD2 with Crohn’s disease in a homogenous Irish population. Eur J Hum Genet 2003;11:237–44. [DOI] [PubMed] [Google Scholar]
- 50.Radlmayr M , Torok HP, Martin K, et al. The c-insertion mutation of the NOD2 gene is associated with fistulizing and fibrostenotic phenotypes in Crohn’s disease. Gastroenterology 2002;122:2091–2. [DOI] [PubMed] [Google Scholar]
- 51.Kurzawski G , Suchy J, Kladny J, et al. The NOD2 3020insC mutation and the risk of colorectal cancer. Cancer Res 2004;64:1604–6. [DOI] [PubMed] [Google Scholar]
- 52.Murillo L , Crusius JB, van Bodegraven AA, et al. CARD15 gene and the classification of Crohn’s disease. Immunogenetics 2002;54:59–61. [DOI] [PubMed] [Google Scholar]
- 53.Mendoza JL, Murillo LS, Fernandez L, et al. Prevalence of mutations of the NOD2/CARD15 gene and relation to phenotype in Spanish patients with Crohn disease. Scand J Gastroenterol 2003;38:1235–40. [DOI] [PubMed] [Google Scholar]
- 54.Vavassori P , Borgiani P, D’Apice MR, et al. 3020insC mutation within the NOD2 gene in Crohn’s disease: frequency and association with clinical pattern in an Italian population. Dig Liver Dis 2002;34:153. [DOI] [PubMed] [Google Scholar]
- 55.Giachino D , Van Duist MM, Regazzoni S, et al. Analysis of the CARD15 variants R702W, G908R and L1007fs in Italian IBD patients. Eur J Hum Genet 2004;12:206–12. [DOI] [PubMed] [Google Scholar]
- 56.Roussomoustakaki M , Koutroubakis I, Vardas EM, et al. NOD2 insertion mutation in a Cretan Crohn’s disease population. Gastroenterology 2003;124:272–3. [DOI] [PubMed] [Google Scholar]
- 57.Croucher PJ, Mascheretti S, Hampe J, et al. Haplotype structure and association to Crohn’s disease of CARD15 mutations in two ethnically divergent populations. Eur J Hum Genet 2003;11:6–16. [DOI] [PubMed] [Google Scholar]
- 58.Vermeire S , Wild G, Kocher K, et al. CARD15 genetic variation in a Quebec population: prevalence, genotype-phenotype relationship, and haplotype structure. Am J Hum Genet 2002;71:74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abreu MT, Taylor KD, Lin YC, et al. Mutations in NOD2 are associated with fibrostenosing disease in patients with Crohn’s disease. Gastroenterology 2002;123:679–88. [DOI] [PubMed] [Google Scholar]
- 60.Aldhous MC, Nimmo ER, Satsangi J. NOD2/CARD15 and the Paneth cell: another piece in the genetic jigsaw of inflammatory bowel disease. Gut 2003;52:1533–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bonen DK, Ogura Y, Nicolae DL, et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 2003;124:140–6. [DOI] [PubMed] [Google Scholar]
- 62.Fellermann K , Wehkamp J, Herrlinger KR, et al. Crohn’s disease: a defensin deficiency syndrome? Eur J Gastroenterol Hepatol 2003;15:627–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.