

Abstract
Background: Helicobacter pylori infection is associated with variable clinical outcomes, including gastroduodenal diseases, and genetic factors may be relevant in this process.
Aims: We investigated the effects of an interleukin 8 (IL-8) gene polymorphism on the risk of gastroduodenal diseases, the degree of H pylori induced gastritis, and IL-8 gene transcription.
Subjects: The study was performed in 244 healthy control subjects and 690 H pylori positive patients with non-cardia gastric cancer, gastric ulcer, duodenal ulcer, or gastritis.
Methods: We identified the IL-8 −251 A/T polymorphism by direct sequence analysis, and measured the gastritis score and serum pepsinogen (PG). The transcriptional promoter activity of the IL-8 gene was assessed by luciferase assay.
Results: IL-8 −251A was associated with a higher risk of gastric cancer and gastric ulcer. Patients carrying IL-8 −251A showed an increased risk of gastric cancer (odds ratios (OR) 2.01 (95% confidence interval (CI) 1.38–2.92)) and gastric ulcer (OR 2.07 (95% CI 1.37–3.12)). Compared with patients younger than 49 years, atrophy and metaplasia scores in the antrum were significantly higher and the PG I/II ratio significantly lower in −251A carriers than in T/T carriers. In the in vitro assay, IL-8 −251A showed enhanced promoter activity in response to IL-1β or tumour necrosis factor α.
Conclusions: The IL-8 −251A allele may be associated with progression of gastric atrophy in patients with H pylori infection, and may increase the risk of gastric cancer and gastric ulcer in Japanese people.
Keywords: interleukin 8, Helicobacter pylori, gastric cancer, gastric atrophy, Japanese, polymorphism
Helicobacter pylori, a bacterium that infects the stomach of humans, is strongly associated with gastroduodenal diseases such as chronic atrophic gastritis,1,2 peptic ulcer,3–5 and gastric cancer.6–8 Although H pylori has been classified as a carcinogen in human,6 the incidence of gastric cancer is paradoxically low in countries such as Nigeria, Thailand, and India in spite of the high prevalence of H pylori infection. This phenomenon has been referred to as the African enigma9 or the Asian enigma.10 From these findings, it is suggested that H pylori infection does not always associate with the risk of gastroduodenal diseases, and some genetic factors of the host are considered to be related to the variety of clinical outcomes induced by H pylori. Recently, some reports have identified polymorphisms of cytokine genes activated by H pylori infection as a host genetic factor. El-Omar et al reported that gene polymorphisms of interleukin 1β (IL-1B) and its receptor antagonist (IL-1RN) were associated with an increased risk of gastric cancer and hypochlorhydria in Caucasian populations from Poland and Scotland.11 Moreover, Machado et al proposed a similar association with gastric cancer in a population from northern Portugal.12 More recently, El-Omar et al reported that the proinflammatory genotypes of tumour necrosis factor α (TNF-α) and IL-10 significantly increased the risk of non-cardia gastric cancer, and that carriage of polymorphisms of multiple proinflammatory cytokines such as IL-1B, IL-1RN, TNF-A, and IL-10 was also associated with a greater risk of gastric cancer.13 However, according to several recent reports in Japanese people,14–16 it is hard to explain the risk of gastric cancer in Japan solely by IL-1B polymorphisms.
Interleukin 8 (IL-8) plays an important role in the pathogenesis of H pylori infection. IL-8, a major host mediator inducing neutrophil chemotaxis and neutrophil activation,17 is produced by gastric epithelial cells as an early response to H pylori infection. IL-8 is also considered to attract and activate phagocytes and cause mucosal damage by releasing reactive oxygen radicals.18 At present, it is suggested that mucosal IL-8 production due to H pylori infection may be an important factor in the immunopathogenesis of peptic ulcer diseases and may also be relevant in gastric carcinogenesis.19
Concerning host genetic factors, Hull et al reported an association between respiratory syncytial virus bronchiolitis and the IL-8 −251A allele in UK families.20 Since the report by Hull et al, others have shown the effects of this allele on inflammatory diseases such as systemic lupus erythematosus nephritis21 and enteroaggregative Escherichia coli diarrhoea.22 However, there has been no report on a possible relation between the IL-8 gene polymorphism (IL-8 −251A/T) and H pylori induced gastroduodenal diseases.
Therefore, the objective of this study was to investigate the effects of IL-8 −251A on the risk of H pylori related gastroduodenal diseases such as gastric cancer and peptic ulcer diseases in Japanese patients. We also wished to assess its association with the degree of H pylori induced gastritis and the serum pepsinogen (PG) I/II ratio, a marker of gastric atrophy.23,24 In addition, we examined the effect of this polymorphism on IL-8 gene transcription activity in a human gastric cancer cell line by luciferase assay.
METHODS
Subjects
We evaluated the IL-8 −251A/T polymorphism in a hospital based case-referent study. The characteristics of the subjects are listed in the table 1 ▶. Subjects in the case group were all Japanese and recruited from inpatients in the gastroenterology section of the University Hospital of Tohoku University School of Medicine (Sendai, Japan) from September 2000 to April 2004. A summary of the study design is shown in fig 1 ▶. A total of 1172 subjects were admitted and underwent endoscopy; the diagnoses were gastric cancer (n = 281), gastric ulcer (n = 272), duodenal ulcer (n = 206), or gastritis (n = 413). All patients were tested for H pylori infection, and 885 H pylori positive patients were enrolled in the study because we focused on the effects of IL-8 −251A on the risk of H pylori related gastric diseases. Patients with cardia cancer or diffuse-type cancer25 were excluded. The histological diagnosis of gastric cancer in this study was entirely the intestinal-type,25 which usually develops on a background of severe atrophic gastritis.26 Patients who had received non-steroidal anti-inflammatory drugs were also excluded, and peptic ulcers caused simply by H pylori were included. Patients were excluded from this study if they refused to be genotyped. A diagnosis of gastritis was based on negative results for macroscopic lesions such as ulcer and cancer but positive for H pylori gastritis by histology or by serology showing antibodies against H pylori. For genotyping, 690 H pylori positive patients (436 men and 254 women; mean age 61.4 years (range 20–87)) with non-cardia gastric cancer (n = 212; located at the corpus and antrum, n = 112 and n = 100, respectively), gastric ulcer (n = 153; located at the corpus and antrum, n = 79 and n = 74, respectively), duodenal ulcer (n = 130), or gastritis (n = 195) were assessed. Among 690 patients, 552 patients were assessed by histological examination and serological evaluation. In 138 subjects, endoscopic biopsy could not be performed due to the presence of concurrent medical conditions.
Table 1.
Characteristics of the subjects in the study
| Control 1 | Control 2 | GC | GU | DU | Gastritis | |
| No of subjects | 244 | 102 | 212 | 153 | 130 | 195 |
| Age (yr) (mean (SD)) | 25.2 (8.2) | 23.5 (6.2) | 68.2 (9.5) | 61.7 (12.3) | 49.7 (16.4) | 61.2 (13.5) |
| Sex (M/F) | 120/124 | 58/44 | 159/53 | 107/46 | 77/53 | 93/102 |
GC, gastric cancer; GU, gastric ulcer; DU, duodenal ulcer.
Figure 1.
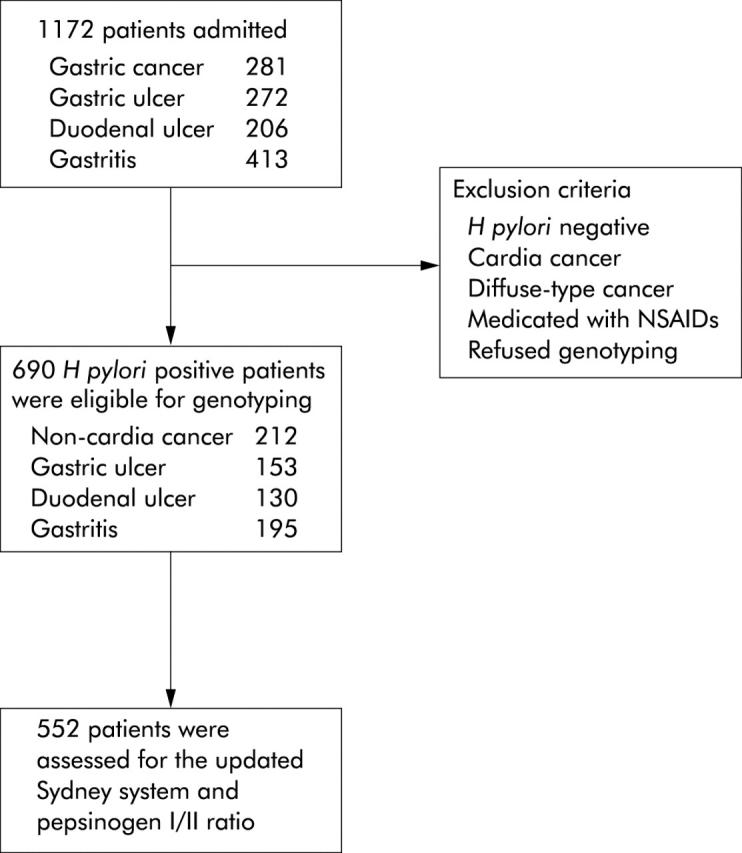
Flow chart of the case groups.
The control group consisted of healthy volunteers with no clinical history of gastroduodenal diseases from among Japanese medical students of Tohoku University School of Medicine. From September 2000 to April 2004, 377 volunteers were enrolled, and 244 subjects were randomly selected and genotyped (control 1). In addition, we used a second set of 102 control individuals (control 2) for further validation of the findings. Informed consent was obtained from all subjects. The study protocol was approved by the Ethics Committee for Human Research of Tohoku University School of Medicine.
DNA extraction and sequencing condition
Genomic DNA was extracted from EDTA anticoagulated peripheral blood leucocytes using a commercially available kit (QIAamp DNA mini kit; Qiagen, Tokyo, Japan). IL-8 −251A/T was genotyped by direct sequence analysis. Firstly, Touchdown polymerase chain reaction (PCR) was carried out. Reaction mixtures consisted of 0.5 μg DNA, 0.25 units Taq polymerase (Takara, Osaka, Japan), 2 mM Tris HCl, pH 8.0, 10 mM KCl, 2 mM MgCl2, 200 μM each of dATP, dTTP, dGTP, and dCTP, and 20 μM each of forward and consensus primers. DNA samples were amplified in a GeneAmp PCR system 9600 (Perkin-Elmer, Foster City, California, USA) with cycling parameters as follows: one minute at 96°C followed by five cycles of 94°C for 30 seconds, 72°C for two minutes, followed by five cycles of 94°C for 30 seconds, 70°C for 20 minutes, followed by 25 cycles of 94°C for 30 seconds, and 68°C for two minutes. The PCR primers were designed based on published IL-8 gene sequences (GenBank accession No: AF385628). For the forward and reverse primers, GCT GGC TTA TCT TCA CCA TCA TGA TAG and GAG CCA CGG CCA GCT TGG AAG TCA TG were used, respectively. The primer set produced a PCR product of 542 bp. This product was sequenced with the ABI Prism Dye Terminator Cycle Sequencing kit (Perkin-Elmer) according to the manufacturer’s instructions using the sequence primer AAC ACC TGC CAC TCT AGT AC, and IL-8 −251A/T was typed using an the ABI 3100 sequencer (Perkin-Elmer). Genotyping of the IL-8 −251 was performed by two independent observers (MO and AI).
Histological examination and serological evaluation
Biopsy specimens were taken from the antrum and corpus along the greater curvature from grossly non-pathological mucosa. These specimens were used for the rapid urease test, bacterial culture, and histological assessment of gastritis. The degrees of inflammation, activity, atrophy, and metaplasia were assessed according to the updated Sydney system,27 and scored from 0 (normal) to 3 (marked). The PG I/ II ratio was calculated based on the data of serum PG I and PG II levels measured by radioimmunoassay. A PG I/II ratio that showed a decrease in proportion to the severity of atrophy23,24 was used as a marker of atrophic gastritis.
H pylori infection was screened by histological examination, the rapid urease test, or antibodies against H pylori. Patients were diagnosed as H pylori positive if one or more of these diagnostic methods was positive.
Luciferase assay
The human gastric cancer cell line AGS was purchased from American Type Culture Collection (Manassas, Virginia, USA) and maintained in medium consisting of F-12 (Invitrogen, Carlsbad, California, USA) supplemented with 10% heat inactivated fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin G, and 100 mg/ml streptomycin.
Firstly, we constructed firefly luciferase reporter gene vectors (pGL3-basic; Promega, Madison, Wisconsin, USA) which included IL-8 promoter regions (−317 to +7) with −251A or T, pGL3-IL8-A, and pGL3-IL8-T. The inserted promoter regions had no mutation when checked by direct sequence analysis. We also used the plasmid pRL-TK (Promega) encoding the Renilla luciferase gene as a background control. For the luciferase assay, AGS was transfected with 2 μg of pGL3-IL8-A or pGL3-IL8-T luciferase vector using FUGENE6 reagent (Roche, Indianapolis, Indiana, USA). After incubation with the vector for 24 hours, IL-1β (final concentration 10 ng/ml; Roche) or TNF-α (final concentration 5 ng/ml; Roche) was added separately. After an additional incubation with IL-1β for eight hours or with TNF-α for 24 hours, luciferase activity in cell extracts was measured using the Dual-Luciferase Reporter Assay System (Promega). Light intensities were measured using a model Lumat LB9507 luminescence reader (EG&G Berthold, Bad Wildbad, Germany). Relative luciferase activity was calculated by normalising luciferase activity to control Renilla luciferase activity, and the relative transcriptional activity was expressed as the percentage of luciferase activity compared with that of non-stimulated cells. We assayed luciferase activity five times independently to obtain high reproducibility and exclude errors.
Statistical analysis
The Hardy-Weinberg equilibrium of the IL-8 gene allele was assessed by χ2 statistics. Differences in the frequency of IL-8 genotypes among gastric cancer, gastric ulcer, duodenal ulcer, or gastritis patients and controls were determined by the χ2 test using a 3×2 contingency table. The individual genotype was analysed using a 2×2 contingency table with Fisher’s exact test, and the odds ratio (OR) with 95% confidence interval (CI) were calculated. Differences in gastritis scores and PG I/ II ratio between the two groups of IL-8 genotypes (A carriers including A/T and A/A, and T/T) were examined by the Mann-Whitney U test. A p value <0.05 was considered statistically significant. The computer program StatView for Windows version 4.54 was used for statistical analysis.
RESULTS
IL-8 −251 genotype
Table 2 ▶ shows IL-8 −251 genotype frequencies in control subjects and H pylori positive patients with gastric cancer, gastric ulcer, duodenal ulcer, or gastritis. The IL-8 −251 genotype of control subjects showed no evidence of deviation from the Hardy-Weinberg equilibrium, with a non-significant χ2 value (χ21 = 0.04, p = 0.85). There were significant differences in genotype distribution between control and gastric cancer (p = 0.0012) or gastric ulcer (p = 0.0007) by the χ2 genotype frequency heterogeneity test using a 3×2 contingency table. The IL-8 −251A allele frequency was higher in subjects with gastric cancer and gastric ulcer compared with controls. To further investigate individual genotype differences, we performed Fisher’s exact test using a 2×2 contingency table. It was estimated that the IL-8 −251 A/T genotype had a significantly higher risk of gastric cancer (OR 2.02 (95% CI 1.37–2.97); p = 0.0005) and gastric ulcer (OR 1.88 (95% CI 1.22–2.89); p = 0.0053) (table 2 ▶). The IL-8 −251 A/A genotype was significantly more frequent in patients with gastric ulcers (OR 3.49 (95% CI 1.55–7.86); p = 0.0033). No significant increase was observed in the risk of gastric cancer for the IL-8 −251 A/A genotype, probably because of the small number of cases with this genotype. When IL-8 −251A/A and A/T were considered together as −251A carriers, their risk of gastric cancer and gastric ulcer was significantly higher (OR 2.01 (95% CI 1.38–2.92), p = 0.0004 and 2.07 (95% CI 1.37–3.12), p = 0.0007), respectively) (table 2 ▶). In contrast, no such association was observed in patients with duodenal ulcer or gastritis. We investigated the IL-8 −251 polymorphism in a second set of control individuals for further validation of the findings (table 3 ▶). A similarly significant correlation was also demonstrated between the second control and cases of gastric cancer or gastric ulcer.
Table 2.
IL-8 −251 genotype frequencies in controls (control 1), and in patients with gastric cancer (GC), gastric ulcer (GU), duodenal ulcer (DU), and gastritis
| Genotype | Control 1 (n = 244) | GC (n = 212) | GU (n = 153) | DU (n = 130) | Gastritis (n = 195) | ||||
| n | OR (95% CI) | n | OR (95% CI) | n | OR (95% CI) | n | OR (95% CI) | ||
| T/T | 149 | 93 | 1.00 | 66 | 1.00 | 68 | 1.00 | 106 | 1.00 |
| A/T | 84 | 106 | 2.02 (1.37–2.97)* | 70 | 1.88 (1.22–2.89)‡ | 57 | 1.49 (0.96–2.31) | 74 | 1.24 (0.83–1.85) |
| A/A | 11 | 13 | 1.89 (0.81–4.40) | 17 | 3.49 (1.55–7.86)§ | 5 | 1.00 (0.33–2.98) | 15 | 1.92 (0.85–4.34) |
| A carrier | 95 | 119 | 2.01 (1.38–2.92)† | 87 | 2.07 (1.37–3.12)¶ | 62 | 1.43 (0.93–2.20) | 89 | 1.32 (0.90–1.93) |
*A/T of GC versus T/T of control: p = 0.0005.
†A carrier of GC versus T/T of control: p = 0.0004.
‡A/T of GU versus T/T of control: p = 0.0053.
§A/A of GU versus T/T of control: p = 0.0033.
¶A carrier of GU versus T/T of control: p = 0.0007.
Table 3.
IL-8 −251 genotype frequencies in controls (control 2), and in patients with gastric cancer (GC), gastric ulcer (GU), duodenal ulcer (DU), and gastritis
| Genotype | Control 2 (n = 102) | GC (n = 212) | GU (n = 153) | DU (n = 130) | Gastritis (n = 195) | ||||
| n | OR (95% CI) | n | OR (95% CI) | n | OR (95% CI) | n | OR (95% CI) | ||
| T/T | 59 | 93 | 1.00 | 66 | 1.00 | 68 | 1.00 | 106 | 1.00 |
| A/T | 34 | 106 | 1.98 (1.19–3.28)* | 70 | 1.84 (1.07–3.16)‡ | 57 | 1.45 (0.84–2.52) | 74 | 1.21 (0.72–2.03) |
| A/A | 9 | 13 | 0.92 (0.37–2.28) | 17 | 1.69 (0.70–4.07) | 5 | 0.48 (0.15–1.52) | 15 | 0.93 (0.38–2.25) |
| A carrier | 43 | 119 | 1.76 (1.09–2.83)† | 87 | 1.81 (1.09–3.00)§ | 62 | 1.25 (0.74–2.11) | 89 | 1.15 (0.71–1.87) |
*A/T of GC versus T/T of control: p = 0.011.
†A carrier of GC versus T/T of control: p = 0.028.
‡A/T of GU versus T/T of control: p = 0.037.
§A carrier of GU versus T/T of control: p = 0.030.
Effects of IL-8 −251 polymorphism on gastritis scores and serological evaluation
When antral atrophy was evaluated according to the ages of the patients it was found that IL-8 −251A carriers showed a significantly higher atrophy score than that of patients with the T/T genotype in the group of patients younger than 49 years (p = 0.028) (table 4 ▶). Moreover, the metaplasia score of the antrum was also substantially increased in the −251A carrier group in this generation compared with the T/T genotype group (p = 0.038) (table 4 ▶). No significant differences in these scores were found in the patients older than 50 years old. Other scores such as inflammation and activity were not significantly different between the −251A carrier and T/T carrier groups (data not shown). The PG I/II ratio showed a tendency to decrease with age in both the −251A carrier and T/T carrier groups, and was lower in −251A carriers in all generations. In particular, the PG I/II ratio was significantly lower in the −251A carrier group than in the T/T group only in patients younger than 49 years old (p = 0.039) (fig 2 ▶).
Table 4.
Atrophy and metaplasia scores of IL-8 −251A carriers and T/T carriers in different generations (⩽49, 50–59, 60–69, and ⩾70 years)
| A carrier | T/T carrier | P Value | |
| ⩽49 years | |||
| n | 64 | 54 | |
| Atrophy | 0.746 (0.883) | 0.449 (0.765) | 0.028 |
| Metaplasia | 0.373 (0.849) | 0.146 (0.540) | 0.038 |
| 50–59 years | |||
| n | 56 | 45 | |
| Atrophy | 1.096 (0.995) | 1.122 (0.954) | 0.779 |
| Metaplasia | 0.538 (0.999) | 0.732 (0.949) | 0.133 |
| 60–69 years | |||
| n | 77 | 79 | |
| Atrophy | 1.181 (0.984) | 1.147 (0.959) | 0.725 |
| Metaplasia | 0.569 (0.853) | 0.697 (0.994) | 0.628 |
| ⩾70 years | |||
| n | 87 | 90 | |
| Atrophy | 1.627 (0.984) | 1.391 (0.932) | 0.168 |
| Metaplasia | 1.289 (1.132) | 1.000 (0.976) | 0.095 |
Atrophy and metaplasia scores are expressed as mean (SD).
Figure 2.
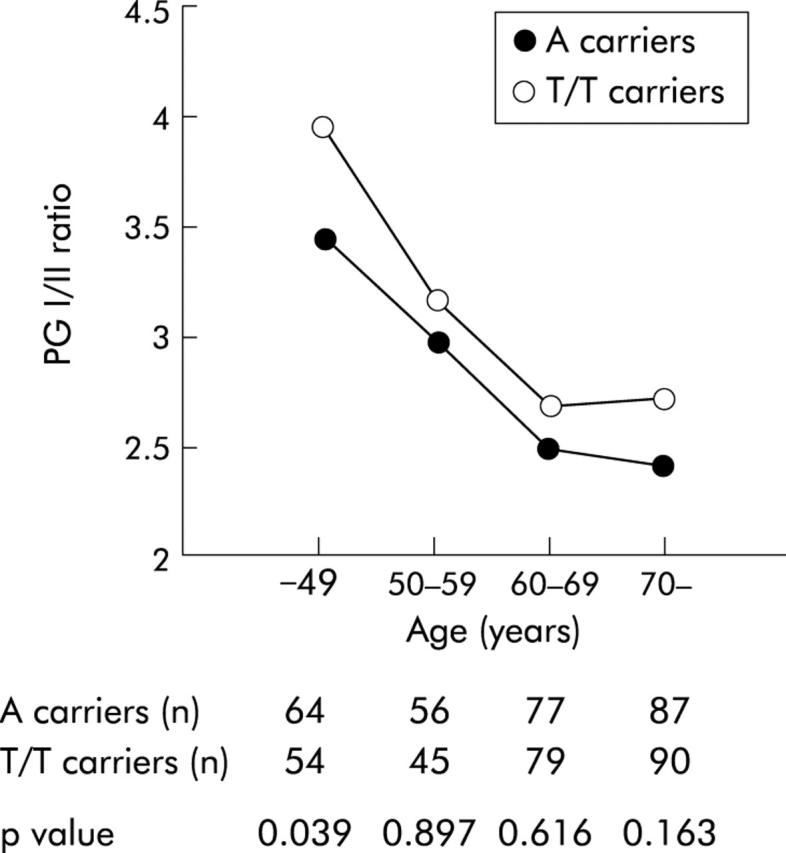
Pepsinogen (PG) I/II ratio of IL-8 −251A carriers and T/T carriers in different generations (⩽49, 50–59, 60–69, and ⩾70 years). In patients younger than 49 years, the PG I/II ratio was significantly lower in −251A carriers than in T/T carriers Data are expressed as medians. Statistical significance was assessed by the Mann-Whitney U test.
Transcriptional promoter activity of the IL-8 gene
We performed the luciferase assay to assess whether IL-8 −251A/T influences the transcriptional promoter activity of the IL-8 gene in response to IL-1β or TNF-α (fig 3 ▶). Relative transcriptional activity was expressed as percentage of luciferase activity compared with unstimulated cells (% rest), as shown in fig 3 ▶. When stimulated with IL-1β, the IL-8 −251A promoter showed significantly higher luciferase activity than the −251T promoter (p = 0.021) (fig 3 ▶). Similarly, TNF-α induced promoter activity was higher in the −251A allele than in the −251T allele although the difference was not statistically significant (p = 0.073) (fig 3 ▶).
Figure 3.

Relationship between the IL-8 −251 single nucleotide polymorphism and the transcriptional promoter activity of the IL-8 gene in response to interleukin 1β (IL-1β 10 ng/ml) or tumour necrosis factor α (TNF-α 5 ng/ml). Relative values were expressed as percentage of luciferase activity compared with non-stimulated cells (% rest). (A) In AGS cells, IL-1β-induced promoter activity of the IL-8 −251A allele (pGL3-IL8-A) was significantly higher than that of the −251T allele (pGL3-IL8-T) (p = 0.021). (B) Similarly, TNF-α induced promoter activity was higher in the −251A allele than in the −251T allele although the difference was not statistically significant (p = 0.073). Values are mean (SD).
DISCUSSION
We have shown that the IL-8 gene polymorphism at position −251 is associated with the risk of gastric cancer and gastric ulcer in Japanese patients with H pylori infection. Allele frequency of IL-8 −251A was higher in patients with gastric cancer or gastric ulcer than in those with duodenal ulcer or gastritis.
Regarding the histopathological differences in gastritis between gastric cancer, gastric ulcer, and duodenal ulcer, Correa has proposed that there are possibly two main pathways of clinical outcomes as a result of H pylori infection.28 One pathway is characterised by diffuse antral gastritis in which the mucosa is infiltrated by lymphocytes and plasma cells. This type of gastritis is typically shown in duodenal ulcer patients29 who are considered to be at no increased risk of gastric cancer.30 The other pathway is characterised by remarkable infiltration of neutrophils with severe damage to the surface epithelium, which may lead to multifocal atrophic gastritis. Some patients with atrophic gastritis develop gastric ulcers, or (and) dysplastic changes associated with gastric cancer. IL-8 is strongly associated with neutrophil activation in H pylori infection. In the present study, we have shown that the promoter activity of IL-8 −251A in response to IL-1β or TNF-α was enhanced compared with −251T. This finding suggested that IL-8 −251A transcription is activated more strongly, leading to more active gastritis with a strong neutrophil infiltration. Considering Correa’s hypothesis, gastritis of IL-8 −251A carriers may lead to atrophic gastritis and the development of gastric ulcers or (and) gastric cancer but is less likely to lead to duodenal ulcers.
With regard to the association between gastric cancer and gastritis, Uemura et al have reported that severe gastric atrophy, corpus predominant gastritis, and intestinal metaplasia are strong risks for the development of gastric cancer.7 Correa et al have also reported that gastric atrophy and hypochlorhydria are strong risk factors for the development of gastric cancer.28,31 On the other hand, Schultze et al reported that antral atrophy and metaplasia were associated with an increased risk of gastric ulcers.32 The aetiological factors of gastric cancer are known to associate positively with gastric ulcers33 and a low PG I/II ratio is a major risk factor for gastric ulcers.34 In particular, Japanese patients with gastric ulcers are considered to present with a higher proportion of atrophy and intestinal metaplasia.35,36 Our findings suggested that IL-8 −251A may be related to the severity of gastric atrophy before 50 years of age because of the more strongly activated IL-8 gene transcription, and may increase the risk of developing gastric atrophy associated diseases such as gastric cancer and gastric ulcers in patients with H pylori infection.
Furthermore, it is well known that a polarised T helper 1 (Th1) immune response occurs in H pylori infection.37 IL-8 production stimulated by H pylori may induce proinflammatory Th1 cytokines such as TNF-α, interferon γ, and IL-1β. Chronic gastritis with a Th1 dominant immunological appearance has been reported to cause gastric atrophy and metaplasia in an H felis infected mouse model.38 Thus a high producer of IL-8 may induce Th1 dominant chronic gastritis which may then be followed by the development of gastric ulcers and gastric cancer. In addition, neutrophils induced by IL-8 synthesise active radicals such as nitric oxide.18 These radicals have mutagenic potential,39 which could cause mutations in gastric epithelial cells. Activated IL-8 gene transcription by IL-8 −251A would induce oxygen radicals, which would be one of the important factors in gastric carcinogenesis.
We have shown that IL-8 −251A is associated with progression of gastric mucosal atrophy. In patients younger than 49 years, the −251A carrier group showed significantly higher scores for antral atrophy and antral metaplasia, and a lower PG I/II ratio, a marker of atrophic gastritis,23,24 than the T/T group. Several studies have shown that long term exposure to H pylori is a significant risk factor for the progression of atrophic gastritis,40–42 hypochlorhydria,43 and intestinal metaplasia.1 In this respect, IL-8 −251A may be considered a factor that promotes gastric mucosal atrophy. In the eldest group of patients, progression of gastric atrophy may be terminated because of the development of complete intestinal metaplasia, possibly because of the influence of long term H pylori infection. Therefore, this may be the reason why there were no significant differences in gastric atrophy between A carriers and T/T carriers in the oldest group of patients. At the same time, we divided subjects into four age groups and performed a relatively large number of statistical tests in which a conventional significance level of p = 0.05 was used without adjustment for multiple comparisons. Such procedures may have increased the likelihood of observing spurious associations for a subgroup of subjects. Therefore, our results in the age specific analyses need to be interpreted cautiously.
Concerning host genetic factors, Thye et al performed a genome wide linkage analysis to identify the genetic factor for the prevalence of H pylori infection, which was defined by serum concentrations of H pylori reactive IgG,44 and suggested the presence of a possible linkage with chromosomes 4 and 6. Indeed, the human IL-8 gene is located on chromosome 4 (4q13–q21). Thus the results of their linkage analysis may support our finding that the IL-8 gene polymorphism is actually associated with H pylori induced gastroduodenal diseases. In the first instance, we investigated whether polymorphisms exist around the 5′-flanking region of IL-8 gene from −650 to +50 by direct sequence analysis of 30 control subjects (data not shown). However, we could not find any polymorphism except for IL-8 −251A/T. Based on this result, we believe that IL-8 −251A is one of the major disease susceptibility factors for gastric cancer and gastric ulcer in Japanese patients.
We did not directly investigate whether gastritis with excess production of IL-8 produces gastric atrophy but it is possible that IL-8 −251A may be one of the major host factors for the progression of gastric mucosal atrophy. The IL-8 promoter contains multiple regulatory transcription factor binding sites. A novel IL-8 expression pathway induced by nickel subsulphide has been reported.45 In this pathway, regulating factors contain binding sites such as GATA (−248 to −245) and C/EBP (−246 to −233). When the allele at position −251 of IL-8 is “A”, the sequence around the −251 region conforms to the binding motif of C/EBP.46 The sequence of the IL-8 promoter region around −251A may contain the binding site of C/EBP. Further studies are necessary to clarify the IL-8 expression pathway through this region.
In conclusion, the present study demonstrated that IL-8 −251A was associated with higher IL-8 gene transcription activity and the development of gastric mucosal atrophy in H pylori infected patients less than 50 years of age, and this allele may increase the risk of gastric cancer and gastric ulcer in the Japanese population. If the IL-8 gene polymorphism shows variation in different ethnic groups, it could be important in explaining the racial differences in the prevalence of H pylori related diseases such as gastric cancer. Further studies on genetic factors are necessary to clarify this issue.
Acknowledgments
We thank Y Kinouchi and K Negoro for advice and expertise. We also thank Y Tsubono for advice on statistical analysis and H Sasano for helpful discussions. Also, we would like to thank Y Abe and M Kawamura for sampling the clinical materials and M Yoshida for technical assistance. The authors are grateful to Brent Bell for reading the manuscript.
Abbreviations
IL, interleukin
IL-1RN, interleukin 1 receptor antagonist
TNF, tumour necrosis factor
PG, pepsinogen
OR, odds ratio
Th1, T helper 1
PCR, polymerase chain reaction
Conflict of interest: None declared.
REFERENCES
- 1.Kuipers EJ, Uyterlinde AM, Pena AS, et al. Long-term sequelae of Helicobacter pylori gastritis. Lancet 1995;345:1525–8. [DOI] [PubMed] [Google Scholar]
- 2.Sipponen P, Kosunen TU, Valle J, et al. Helicobacter pylori infection and chronic gastritis in gastric cancer. J Clin Pathol 1992;45:319–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodwin CS, Mendall MM, Northfield TC. Helicobacter pylori infection. Lancet 1997;349:265–9. [DOI] [PubMed] [Google Scholar]
- 4.Hopkins RJ, Girardi LS, Turney EA. Relationship between Helicobacter pylori eradication and reduced duodenal and gastric ulcer recurrence: a review. Gastroenterology 1996;110:1244–52. [DOI] [PubMed] [Google Scholar]
- 5.Graham DY, Lew GM, Klein PD, et al. Effect of treatment of Helicobacter pylori infection on the long-term recurrence of gastric or duodenal ulcer. A randomized, controlled study. Ann Intern Med 1992;116:705–8. [DOI] [PubMed] [Google Scholar]
- 6.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Schistosomes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 7.Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001;345:784–9. [DOI] [PubMed] [Google Scholar]
- 8.Hansson LE, Engstrand L, Nyren O, et al. Helicobacter pylori infection: independent risk indicator of gastric adenocarcinoma. Gastroenterology 1993;105:1098–103. [DOI] [PubMed] [Google Scholar]
- 9.Holcombe C . Helicobacter pylori: the African enigma. Gut 1992;33:429–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miwa H, Go MF, Sato N. H. pylori and gastric cancer: the Asian enigma, Am J Gastroenterol 2002;97:1106–12. [DOI] [PubMed] [Google Scholar]
- 11.El-Omar EM, Carrington M, Chow WH, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000;404:398–402. [DOI] [PubMed] [Google Scholar]
- 12.Machado JC, Pharoah P, Sousa S, et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001;121:823–9. [DOI] [PubMed] [Google Scholar]
- 13.El-Omar EM, Rabkin CS, Gammon MD, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003;124:1193–201. [DOI] [PubMed] [Google Scholar]
- 14.Furuta T, El-Omar EM, Xiao F, et al. Interleukin 1beta polymorphisms increase risk of hypochlorhydria and atrophic gastritis and reduce risk of duodenal ulcer recurrence in Japan. Gastroenterology 2002;123:92–105. [DOI] [PubMed] [Google Scholar]
- 15.Kato S, Onda M, Yamada S, et al. Association of the interleukin-1 beta genetic polymorphism and gastric cancer risk in Japanese. J Gastroenterol 2001;36:696–9. [DOI] [PubMed] [Google Scholar]
- 16.Hamajima N, Matsuo K, Saito T, et al. Interleukin 1 polymorphisms, lifestyle factors, and Helicobacter pylori infection. Jpn J Cancer Res 2001;92:383–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Craig PM, Territo MC, Karnes WE, et al. Helicobacter pylori secretes a chemotactic factor for monocytes and neutrophils. Gut 1992;33:1020–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang QB, Dawodu JB, Husain A, et al. Association of antral mucosal levels of interleukin 8 and reactive oxygen radicals in patients infected with Helicobacter pylori. Clin Sci 1997;92:69–73. [DOI] [PubMed] [Google Scholar]
- 19.Crabtree JE, Lindley IJ. Mucosal interleukin-8 and Helicobacter pylori-associated gastroduodenal disease. Eur J Gastroenterol Hepatol 1994;6 (suppl 1) :S33–8. [PubMed] [Google Scholar]
- 20.Hull J, Thomson A, Kwiatkowski D. Association of respiratory syncytial virus bronchiolitis with the interleukin 8 gene region in UK families. Thorax 2000;55:1023–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rovin BH, Lu L, Zhang X. A novel interleukin-8 polymorphism is associated with severe systemic lupus erythematosus nephritis. Kidney Int 2002;62:261–5. [DOI] [PubMed] [Google Scholar]
- 22.Jiang ZD, Okhuysen PC, Guo DC, et al. Genetic susceptibility to enteroaggregative Escherichia coli diarrhea: polymorphism in the interleukin-8 promotor region. J Infect Dis 2003;188:506–11. [DOI] [PubMed] [Google Scholar]
- 23.Asaka M, Kimura T, Kudo M, et al. Relationship of Helicobacter pylori to serum pepsinogens in an asymptomatic Japanese population. Gastroenterology 1992;102:760–6. [DOI] [PubMed] [Google Scholar]
- 24.Kekki M, Samloff IM, Varis K, et al. Serum pepsinogen I and serum gastrin in the screening of severe atrophic corpus gastritis. Scand J Gastroenterol Suppl 1991;186:109–16. [DOI] [PubMed] [Google Scholar]
- 25.Lauren P . The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma: an attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 1965;64:31–49. [DOI] [PubMed] [Google Scholar]
- 26.Correa P . Human gastric carcinogenesis: a multistep and multifactorial process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res 1992;52:6735–40. [PubMed] [Google Scholar]
- 27.Dixon MF, Genta RM, Yardley JH, et al. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol 1996;20:1161–81. [DOI] [PubMed] [Google Scholar]
- 28.Correa P . Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995;19:S37–43. [PubMed] [Google Scholar]
- 29.El-Omar EM, Penman ID, Ardill JE, et al. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology 1995;109:681–91. [DOI] [PubMed] [Google Scholar]
- 30.Lee S, Iida M, Yao T, et al. Risk of gastric cancer in patients with non-surgically treated peptic ulcer. Scand J Gastroenterol 1990;25:1223–6. [DOI] [PubMed] [Google Scholar]
- 31.Correa P, Haenszel W, Cuello C, et al. A model for gastric cancer epidemiology. Lancet 1975;2:58–60. [DOI] [PubMed] [Google Scholar]
- 32.Schultze V, Hackelsberger A, Gunther T, et al. Differing patterns of Helicobacter pylori gastritis in patients with duodenal, prepyloric, and gastric ulcer disease. Scand J Gastroenterol 1998;33:137–42. [DOI] [PubMed] [Google Scholar]
- 33.Hansson LE, Nyren O, Hsing AW, et al. The risk of stomach cancer in patients with gastric or duodenal ulcer disease. N Engl J Med 1996;335:242–9. [DOI] [PubMed] [Google Scholar]
- 34.Samloff IM, Stemmermann GN, Heilbrun LK, et al. Elevated serum pepsinogen I and II levels differ as risk factors for duodenal ulcer and gastric ulcer. Gastroenterology 1986;90:570–6. [DOI] [PubMed] [Google Scholar]
- 35.Unge P, Kimura K, Sipponen P, et al. Do Japanese and Swedish peptic ulcer patients respond differently to Helicobacter pylori eradication therapies and what are their histological features? Scand J Gastroenterol 2003;38:482–90. [DOI] [PubMed] [Google Scholar]
- 36.Tatsuta M, Iishi H, Ichii M, et al. Chromoendoscopic observations on extension and development of fundal gastritis and intestinal metaplasia. Gastroenterology 1985;88:70–4. [DOI] [PubMed] [Google Scholar]
- 37.Bamford KB, Fan X, Crowe SE, et al. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 1998;114:482–92. [DOI] [PubMed] [Google Scholar]
- 38.Fox JG, Beck P, Dangler CA, et al. Concurrent enteric helminth infection modulates inflammation and gastric immune responses and reduces helicobacter-induced gastric atrophy. Nat Med 2000;6:536–42. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen T, Brunson D, Crespi CL, et al. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Sci U S A 1992;89:3030–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ihamaki T, Kekki M, Sipponen P, et al. The sequelae and course of chronic gastritis during a 30- to 34-year bioptic follow-up study. Scand J Gastroenterol 1985;20:485–91. [DOI] [PubMed] [Google Scholar]
- 41.Kimura K . Chronological transition of the fundic-pyloric border determined by stepwise biopsy of the lesser and greater curvatures of the stomach. Gastroenterology 1972;63:584–92. [PubMed] [Google Scholar]
- 42.Kawaguchi H, Haruma K, Komoto K, et al. Helicobacter pylori infection is the major risk factor for atrophic gastritis. Am J Gastroenterol 1996;91:959–62. [PubMed] [Google Scholar]
- 43.Haruma K, Kamada T, Kawaguchi H, et al. Effect of age and Helicobacter pylori infection on gastric acid secretion. J Gastroenterol Hepatol 2000;15:277–83. [DOI] [PubMed] [Google Scholar]
- 44.Thye T, Burchard GD, Nilius M, et al. Genomewide linkage analysis identifies polymorphism in the human interferon-gamma receptor affecting Helicobacter pylori infection. Am J Hum Genet 2003;72:448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barchowsky A, Soucy NV, O’Hara KA, et al. A novel pathway for nickel-induced interleukin-8 expression. J Biol Chem 2002;277:24225–31. [DOI] [PubMed] [Google Scholar]
- 46.Osada S, Yamamoto H, Nishihara T, et al. DNA binding specificity of the CCAAT/enhancer-binding protein transcription factor family. J Biol Chem 1996;271:3891–6. [DOI] [PubMed] [Google Scholar]