

Abstract
Chromosomes are replicated in characteristic, temporal patterns during S phase. We have compared the timing of association of replication proteins at early- and late-replicating origins of replication. Minichromosome maintenance proteins assemble simultaneously at early- and late-replicating origins. In contrast, Cdc45p association with late origins is delayed relative to early origins. DNA polymerase α association is similarly delayed at late origins and requires Cdc45p function. Activation of the S phase checkpoint inhibits association of Cdc45p with late-firing origins. These studies suggest that Cdc45p is poised to serve as a key regulatory target for both the temporal and checkpoint-mediated regulation of replication origins.
The replication of an eukaryotic chromosome is a complex, highly regulated process. DNA replication initiates from multiple origins on each chromosome. The chromosomal location of each origin is determined by cis-acting sequences termed replicators (1, 2). By strictly regulating replicator activity, cell cycle regulatory proteins limit DNA replication to a single, complete round during each S phase (3). Within S phase, chromosomal position influences replication timing, establishing characteristic, temporal patterns of genome duplication (4). Chromosomal position effects causing late replication and transcriptional silencing have been associated with condensed chromatin structures termed heterochromatin (5). Although the cause and effect relationship between replication timing, gene expression, and chromatin structure remains obscure, heterochromatin may regulate the timing of DNA replication and gene expression through a common mechanism. Heterochromatic gene silencing is thought to occur by restricting the accessibility of transactivators to their target promoters (6, 7). Similarly, heterochromatin may temporally regulate the accessibility of one or more replication factors to replicators.
In Saccharomyces cerevisiae, chromosomal replicators were initially identified as DNA sequences that promoted the replication of episomal DNA and were thus referred to as autonomously replicating sequences (ARS) (8, 9). On plasmids, most ARS elements initiate replication early in S phase; however, within certain chromosomal contexts, such as telomeric regions, ARS elements replicate late in S phase (10). The late replication of these ARS elements and surrounding chromosomal regions is not caused by inherent sequence differences in the ARS elements but depends on the nature of the flanking DNA sequences (11, 12). Cell cycle experiments indicate that the chromosomal position effect on replication timing is established within M phase or early G1 phase (13). Taken together, these results suggest that a chromatin structure assembled during M or early G1 phase regulates origin activity. Understanding the molecular events that distinguish early- and late-replicating origins should provide insights into the mechanisms that regulate replication timing.
Recent experiments using chromatin immunoprecipitation and chromatin fractionation procedures have permitted the identification of factors and characterization of their assembly into replication complexes at origins (14). Biochemically distinct replication complexes assemble in a step-wise manner through a cell-cycle regulated process. The first step in this process is the recognition of and binding to replication origins by the origin recognition complex. The origin recognition complex appears to be origin-associated throughout the cell cycle (15–17). As cells exit mitosis and enter G1 phase, Cdc6p associates with origins through an origin recognition complex-dependent mechanism. The origin recognition complex and Cdc6p are in turn required for association in G1 phase of minichromosome maintenance (MCM) proteins to form the pre-replicative complex (pre-RC) (16, 17). S-phase cyclin-dependent kinases (S-CDKs) and/or the Cdc7p/Dbf4p kinase are thought to drive S-phase entry by regulating the activity of one or more pre-RC components (18). Cdc45p may represent such a target. Whereas association of Cdc45p with the pre-RC in G1 phase has been observed (16), its stable association with total chromatin correlates with activation of S-CDKs at the G1/S transition (19). The initiation of DNA replication leads to a rearrangement of pre-RC components and recruitment of DNA polymerases to assemble the replicative complex (RC) (16). This transition involves dissociation of Cdc6p from the pre-RC (17) as the MCM proteins and Cdc45p assemble into the RC (16). It is not known how the MCM proteins and Cdc45p function in replication, which steps in the replication process S-CDKs and Cdc7p/Dbf4p stimulate, or which steps are differentially regulated at early- and late-replicating origins.
Because pre-RCs are assembled in early G1 phase at or after the time when cell cycle experiments indicate that replication timing is determined, it is likely that assembly of one or more factors into the pre-RC is distinct at early versus late origins. To identify such factors, we compared the kinetics of pre-RC and RC assembly at early- and late-replicating origins. Association of Cdc45p and DNA polymerase α with late-replicating origins was delayed relative to early-replicating origins. Consistent with Cdc45p mediating this delayed polymerase α association, we present evidence that Cdc45p activity is required for polymerase loading onto replication origins. Furthermore, we show that activation of the S phase checkpoint inhibits the association of Cdc45p with late-firing replication origins. These findings suggest that Cdc45p is a key regulatory target of the factors that regulate the temporal program of chromosomal DNA replication and checkpoint surveillance.
MATERIALS AND METHODS
Plasmid and Strain Constructions.
A triple-HA-epitope-tagged gene fusion of CDC17 (Polα) was created as described for CDC54 (Mcm4p), CDC47 (Mcm7p), CDC45 (Cdc45p), and POL2 (Polɛ) (16). In brief, the C-terminal coding region of CDC17 was PCR amplified and inserted into pSF323. A SacI-triple-HA cassette was inserted to create a C-terminal fusion with the coding sequence. This C-terminal, triple-HA-tagged gene fusion was inserted into pRS404 (20), yielding p404-CDC17-HA/C. Plasmid p306-CDC45-HA/C was constructed by inserting the 1.9-kbp KpnI-SacI fragment containing the triple-HA-tagged CDC45 C terminus (from p404-CDC45-HA/C) into pRS306.
All yeast strains are congenic and haploid, bar1∷hisG derivatives of W303–1a. Strains OAy470, OAy534, OAy617, OAy618, and OAy556 and the plasmids used in their construction have been described (16). OAy658 was constructed by transformation of OAy550 (cdc15–2) with linearized p306-CDC45-HA/C. OAy644, OAy676, and OAy679 were constructed by transformation of OAy470 (wild-type), DLy259 (rad53–11), and OAy657 (cdc45–1), respectively, with linearized p404-CDC17-HA/C. OAy664 was constructed by transformation of OAy657 with linearized p405-Polɛ-HA/C. OAy684 was constructed by transformation of DLy259 (rad53–11) with linearized p404-CDC45-HA/C. Epitope tags were constructed by homologous recombination that deleted the wild-type allele while simultaneously creating a C-terminally epitope-tagged version of the gene.
Yeast Methods.
Synchronization methods have been described (16) with minor modifications: Phthalic acid was not used for α factor arrests, and cdc15–2 cells were blocked by incubation at 37°C for 2 hours. In the experiments in Fig. 1, cells were released from the G1 phase block at 16°C to increase the temporal resolution of the assay. Fluorescence-activated cell sorting (FACS) analysis was performed as described except that incubations with RNase A and Proteinase K were extended to at least 2 hours each (21). Chromatin immunoprecipitations were performed as described with anti-HA monoclonal antibody (12CA5) (22).
Figure 1.

Temporally regulated origin associations of Mcm7p, Cdc45p, and DNA polymerases α and ɛ. (A–D) Cells of strains OAy534 [Mcm7p-HA (A)], OAy617 [Cdc45p-HA (B)], OAy644 [Polα-HA (C)], and OAy618 [Polɛ-HA (D)] were synchronized in G1 phase with α factor and were released at 16°C (t = 0 min). At the indicated time points, chromatin-containing extracts were prepared from formaldehyde cross-linked cells and were immunoprecipitated with anti-HA monoclonal antibody. PCR with primers specific to the ARS305, ARS1, ARS501, and ARS603 chromosomal replication origins and the non-origin sequences 305+8kb and R11 amplified the precipitated DNA for analysis by PAGE and EtBr staining. (E) FACS analysis indicates the DNA content of cells throughout the time course in B. FACS profiles from the experiments in A, C, and D were very similar. (F) Budding index (% Unbudded) of cells throughout each time course in A–D was determined and plotted.
PCR Analysis and Primers.
Of the precipitated DNA, 1/50th was subjected to 28 PCR cycles under previously described conditions, and the products were analyzed by PAGE and EtBr staining (16). Amplification of “Input” DNA samples and quantification methods were performed as described except that densitometry was performed directly on negative images of the gels (16). Sequences of the primers used are ARS305-1, 5′-ctccgtttttagccccccgtg-3′; ARS305-2, 5′-gattgaggccacagcaagaccg-3′; ARS306-1, 5′-gcaagcatcttgtttgtaacgcga-3′; ARS306-2, 5′-cctcagcgatgtgctcgctc-3′; ARS606-1, 5′-ggtcttcttgataattctgtggg-3′; ARS606-2, 5′-gaaaaagagattaattgtaggaaagagg-3′; ARS607-1, 5′-cagagaaactattacattgtacattctcc-3′; ARS607-2, 5′-tgtgccgaataatgtgtaagtctc-3′; ARS1-1, 5′-ggtgaaatggtaaaagtcaaccccctgcg-3′; ARS1-2, 5′-gctggtggactgacgccagaaaatgtt-3′; ARS501-1, 5′-cttttttaatgaagatgacattgctcc-3′; ARS501-2, 5′-gatgatgatgaggagctccaatc-3′; ARS603-1, 5′-ctctttcccagatgatatctagatgg-3′; ARS603-2, 5′-cgaggctaaattagaatttttgaagtc-3′; 305+8kb1, 5′-ggtggtggagaagcggttcaaag-3′; 305+8kb, 5′-ccgctcgtacccgctcctga-3′; R11-1, 5′-caccgatacgtacttaaactcttccg-3′; R11-2, 5′-gagaaagcttagtccattcggcc-3′.
RESULTS
Temporal Regulation of pre-RC and RC Assembly.
We have analyzed the association of three MCM proteins (Mcm3p, Mcm4p, and Mcm7p), Cdc45p, DNA polymerase α (Polα), and DNA polymerase ɛ (Polɛ) with early- and late-replicating chromosomal origins of replication by using a chromatin immunoprecipitation procedure (23). In most experiments, we tested for coprecipitation of origin DNA from the early-replicating origins, ARS305 and ARS1, and the late-replicating origins, ARS501 and ARS603 (10, 41–43). Additionally, two non-origin DNA sequences were used to monitor the origin-specificity of protein–DNA interactions as well as replication fork movement; 305+8kb lies 8 kbp from the ARS305 origin and replicates early whereas R11 lies 16 kbp from the ARS501 origin and replicates late (10, 41). DNA sequences were judged to be associated with the epitope-tagged protein of interest when the coprecipitated sequence was enriched relative to the same sequence from an untagged strain (ref. 16; data not shown). Throughout this work, results with Mcm3p, Mcm4p, and Mcm7p were indistinguishable.
In cells blocked in G1 phase with α-factor, Mcm7p and Cdc45p were associated with the early-replicating origins ARS305 and ARS1 whereas neither DNA polymerase α nor ɛ had loaded onto early origin sequences in G1 phase (Figs. 1 and 2, t = 0 min). On release (at 16°C) from the G1 arrest into the cell cycle, Polα and Polɛ loaded concurrently onto the early-replicating origins with the peak signal occurring before bulk DNA replication (based on FACS analysis) and bud emergence (Figs. 1 C–F and 2 C and D, t = 48–60 min). The Cdc45p-early origin associations also exhibited signal increases that coincided with the peak signal observed for the polymerases with the early origins (Figs. 1 and 2, t = 48–60 min). It is not clear whether this peak reflects an increase in the number of protein–origin complexes in the cell, a change in protein stoichiometry, or a change in the architecture of the origin complex that increased crosslinking efficiency. Dissociation of Mcm7p, Cdc45p, Polα, and Polɛ from the early origins coincided with initial bulk DNA synthesis and bud emergence (Figs. 1 and 2, t = 72–84 min). The dissociation of these proteins from ARS305 was accompanied by their detection at a non-origin DNA sequence 8 kbp away from ARS305 (305+8kb).
Figure 2.

Quantification of the data for ARS305 and ARS603 in Fig. 1 A–D is plotted in A, B, C, and D, respectively. % Precipitated, determined as the densitometric value for each PCR product of an immunoprecipitated sample divided by the value for the PCR reaction of the corresponding input DNA sample multiplied by the appropriate dilution factor.
Although MCM protein assembly was comparable at early and late origins, the timing of association of other factors with these two classes of origins clearly differed (Figs. 1 and 2). In contrast to the early origins described above, Cdc45p, Polα, and Polɛ showed only a background signal with late-replicating origins in G1 phase, equivalent to the signal obtained with an untagged strain (data not shown). Instead, these factors did not show significant association with late origins until S phase (Figs. 1 and 2, t = 84–96 min, and FACS). Dissociation of Mcm7p, Cdc45p, Polα, and Polɛ from the late origins occurred as the bulk of DNA synthesis was completed, and was accompanied by the detection of these proteins at a non-origin DNA sequence 16 kbp away from ARS501 (R11). These results show that Cdc45p and the polymerases bind to late-replicating origins substantially later in the cell cycle than to their early-replicating counterparts. Because Cdc45p is required for initiation of replication and Polα is required for initial DNA synthesis from the RNA-primed template (24, 25), these results provide strong evidence that late origins are blocked at a stage of protein assembly before initiation of replication rather than at DNA synthesis or replication fork movement.
Because the MCM proteins had already bound to pre-RCs in G1 blocked cells (Figs. 1A and 2A), it remained possible that the time of MCM proteins binding as cells entered G1 phase was distinct at early versus late origins. To address this possibility, we examined the kinetics of MCM association after release (at 23°C) from a cell cycle block in late mitosis (cdc15–2) when MCM proteins were not yet chromatin-associated. Here, we analyzed Mcm4p-origin association at three additional early-firing origins, ARS306, ARS607, and ARS606 (41–43). The timing of Mcm4p association with origins in early G1 phase was indistinguishable between the early- and late-replicating origins (Fig. 3A; for quantification, see Fig. 6, which is published as supplemental data on the PNAS web site, www.pnas.org).
Figure 3.

Cdc45p associates with early-replicating origins in G1 phase. Haploid cells of strains OAy556 [cdc15–2, Mcm4p-HA (A)] and OAy658 [cdc15–2, Cdc45p-HA (B and C)] were synchronized in late M phase by incubating at 37°C for 2 hours and were released at 23°C (t = 0 min). To block cells in G1 phase, α factor was added 15 min before release from the M phase arrest in C. At 12-min (A and B) or 24-min (C) intervals, samples were fixed for chromatin immunoprecipitations with anti-HA antibody, FACS analysis, and budding index determination. In addition to the origin sequences analyzed in Fig. 1, ARS306, ARS607, and ARS606 were analyzed by PCR. Quantification of the ARS305 and ARS603 data in A and B and all of the data in C is plotted in Fig. 6, which is published as supplemental data on the PNAS web site, www.pnas.org. Because blocked-and-released cdc15 cells are delayed in cytokinesis, unseparated cells with new (small) buds were scored as two small-budded cells (or one small-budded cell and one unbudded cell when only one bud was visible on the unseparated pair of cells). The delay in cytokinesis also resulted in an apparent 2C-to-4C shift in DNA content as replication proceeded.
Cdc45p-Origin Association in G1 Phase Correlates with Early Origin Activation.
Similar to the studies with G1 blocked and released cells, the timing of Cdc45p-origin association was distinct at early- and late-replicating origins released from a mitotic block. After release from a late M phase block (cdc15–2), the relative timing of Cdc45p-origin association again correlates with the relative timing of origin activation. Cdc45p first associated with the early origins just before DNA synthesis (Fig. 3B, t = 36 min; for quantification, see supplemental data at www.pnas.org) but associated with the late origins when ≈50% of chromosomal DNA was replicated (Fig. 2B, t = 60 min). The peak association of Cdc45p relative to Mcm4p was delayed by one time point (12 min) at the early origins and by three time points (36 min) at the late origins. To confirm that the association of Cdc45p with the early origins was occurring in G1 phase, we repeated the M phase block and release experiment in the presence of α factor to prevent entry into S phase. Cdc45p associated with four of the five early origins tested but was not observed at the late-firing origins in G1 blocked cells (Fig. 3C; note the lack of DNA replication by FACS and the lack of new bud emergence). We have previously demonstrated association of Cdc45p with ARS1 in G1 phase cells (Fig. 1 and ref. 16) and suspect the lack of Cdc45p-ARS1 association in this experiment may be attributable to the M phase block combined with a prolonged G1 arrest that results in reduced Cdc45p levels (26). Furthermore, in G1-arrested cells, association of Cdc45p with ARS1 is abolished by mutations in conserved sequence elements of ARS1 required for origin function (O.M.A. and S.P.B., unpublished work). These results provide clear evidence that Cdc45p specifically associates with early but not late origins before the initiation of DNA replication.
Origin-Loading of DNA Polymerase α and DNA Polymerase ɛ Requires Cdc45p.
Polymerase α and ɛ loading occurs subsequent to Cdc45p binding at early origins and with very similar timing as Cdc45p at late origins (Figs. 1 and 2). To determine whether origin-loading of DNA polymerase α or ɛ requires Cdc45p activity, we monitored Polα- or Polɛ-loading in wild-type cells and cells with a defective allele of CDC45 [cdc45–1 (27)] during a synchronous release into the cell cycle from a G1 phase block. In wild-type cells at 12°C, both polymerases loaded onto early origins before bulk DNA replication and loaded onto late-replicating origins in mid-S phase (Fig. 4; data not shown for Polɛ). However, in cdc45–1 mutant cells at the nonpermissive temperature (12°C), neither Polα nor Polɛ origin-loading was detected, and little or no bulk DNA replication was observed. In the cdc45–1 mutant cells at the nonpermissive temperature, MCM proteins maintained origin association (data not shown). These data demonstrate that Cdc45p function is required to load DNA polymerases α and ɛ onto replication origins.
Figure 4.

Origin-loading of DNA polymerase α requires Cdc45p function. Cells of strains OAy644 (WT, Polα-HA) and OAy679 (cdc45–1, Polα-HA) were synchronized in G1 phase with α factor and were released at 12°C (t = 0 min). Samples were collected at 30-min intervals (no samples collected at t = 30 or 60 min) for chromatin immunoprecipitation analysis, FACS analysis, and budding index determination (% Unbudded). PCR analysis was performed as in Fig. 1.
The S Phase Checkpoint Regulates Cdc45p Origin Association.
DNA damage or blocks to replication elongation elicit a cell cycle checkpoint response that blocks progression through mitosis (28). Rad53p is a key mediator of this checkpoint pathway and mutations of RAD53 that inactivate the checkpoint response result in lethality of cells exposed to DNA replication inhibitors such as hydroxyurea or DNA damaging agents. Recent studies have shown that late origin activation is inhibited in cells released into S phase in the presence of hydroxyurea or MMS whereas early origin firing is unaffected; the inhibition of late origin firing depends on the Rad53p checkpoint pathway (29, 30). This finding suggests that Rad53p regulates a component of the origin complex thus placing (late) origin activation under checkpoint surveillance. Because Cdc45p is present at early- but not late-firing pre-RCs, we tested whether association of Cdc45p with origin DNA was inhibited by hydroxyurea treatment.
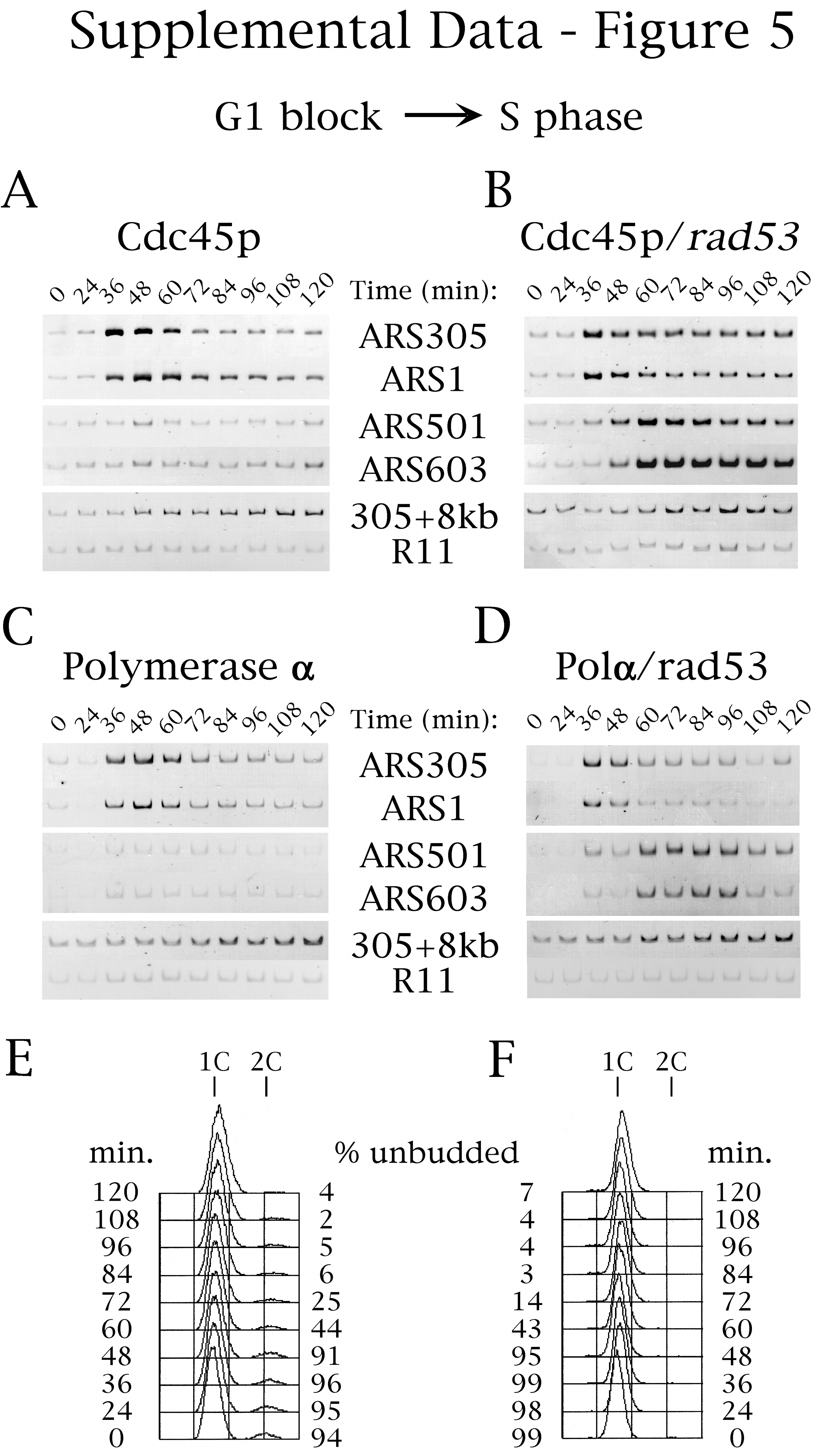
Hydroxyurea inhibited Cdc45p association with late-replicating origins in wild-type but not in rad53 mutant cells (Fig. 5 A and B; see supplemental Fig. 7 at www.pnas.org for gel data including ARS1 and ARS501 and for FACS analysis). Similarly, DNA Polα loading at late origins was inhibited by hydroxyurea (and MMS) in wild-type but not in rad53 mutant cells (Fig. 5 C and D; data not shown for MMS), presumably because of the dependence of Polα-origin loading on Cdc45p (Fig. 4). Because Mcm4p and Mcm7p maintained their associations with the late origins in the presence of hydroxyurea (data not shown), the above results argue that Rad53p acts to inhibit late origin firing by directly or indirectly blocking Cdc45p-origin association, and this regulation is sufficient to explain the block to late origin activation.
Figure 5.

Rad53p regulates Cdc45p binding to late-firing origins. Cells of strains OAy617 [Wild-type, Cdc45p-HA (A)], OAy684 [rad53, Cdc45p-HA (B)], OAy644 [Wild-type, Polα-HA (C)], and OAy676 [rad53, Polα-HA (D)] were synchronized in G1 phase with α factor and were released at 23°C into medium containing hydroxyurea (200 mM) (t = 0 min). Every 12 min, samples were removed for chromatin immunoprecipitation analysis, FACS analysis, and budding index determination (% Unbudded). PCR analysis was performed as in Fig. 1. The quantified data is presented. The original gels including all of the primer data are shown in Fig. 7, which is published as supplemental data at www.pnas.org. In addition, the FACS analyses of the experiments shown in A and B are available as supplemental data (Fig. 7 E and F, respectively). The failure to detect Cdc45p-origin association in G1 cells treated with hydroxyurea was likely attributable to reaction of formaldehyde with the hydroxyurea amine groups, quenching the level of crosslinking below that necessary for detection of Cdc45p association with G1 phase origins.
DISCUSSION
We have investigated the molecular basis for differences in replication timing along eukaryotic chromosomes by analyzing the origin-association kinetics of MCM proteins, Cdc45p, DNA polymerase α, and DNA polymerase ɛ. Our analysis reveals differences in the cell cycle associations of Cdc45p and DNA polymerases with early and late origins of replication. In particular, we demonstrate that Cdc45p associates with early origins in G1 but does not bind to late-firing origins until mid-S phase, when late origins initiate replication. Moreover, our evidence argues that association of Cdc45p with origins is required for DNA polymerases to load at these same sites. These findings indicate that assembly of replication complexes at early and late origins is distinct before replication initiation.
A recent study showed a delay in Cdc45p association with total chromatin in the absence of S-CDKs (Δclb5 and Δclb6), suggesting that S-CDKs are required for Cdc45p-origin association (19). This would appear to conflict with our data showing that Cdc45p binds specifically to early-firing origins during G1 before activation of S-CDKs (Figs. 1B, 2B, and 3C). The difference in these results is likely attributable to the different methods used to assess Cdc45p association with chromatin and may reflect distinct modes of Cdc45p-chromatin association, which we term “bound” (CDK-independent) and “engaged” (CDK-dependent) (31). Crosslinking may stabilize Cdc45p association with G1 phase origins (bound) that is otherwise disrupted during isolation of noncrosslinked chromatin. Possibly, the CDK-dependent chromatin-association of Cdc45p reflects a modification that stabilizes Cdc45p-origin interactions such as the incorporation of Cdc45p into the RC (engaged). Such a change in the interaction of Cdc45p with chromatin may contribute to the enhanced signal we observe during initiation of replication (Figs. 1B and 2B, t = 60 min).
Cdc45p appears to participate in an early step in the initiation process. We have shown that Cdc45p association with early origins occurs before replication initiation, and the recruitment of DNA polymerases to origins requires Cdc45p activity. Therefore, regulation of Cdc45p-origin association provides a mechanism to delay origin firing. For example, a Cdc45p-containing pre-RC may be the required substrate for origin activation by either S-CDKs (Clb5p-Cdk1p and Clb6p-Cdk1p) or Cdc7p/Dbf4p. Clb5p-Cdk1p is required for activation of late-replicating origins and, as indicated above, may act to engage Cdc45p onto the origin complex (19, 32). Cdc7p is also required to promote late origin initiation and appears to function subsequent to S-CDKs (33–35). In addition, reciprocal shift experiments suggest Cdc45p and Cdc7p/Dbf4p function at the same step during the initiation of DNA replication (26). Thus, engagement of Cdc45p at origins by S-CDKs may provide the substrate required for Cdc7p/Dbf4p to initiate replication, consistent with data indicating that chromatin association of Cdc45p is independent of Cdc7p function (19).
Understanding how Cdc45p association with late origins is delayed should reveal important clues about how replication timing is regulated. Late replication of ARS501 is programmed during the preceding M or early G1 period of the cell cycle (13). The molecular basis for establishing the late replication program is unknown but may involve assembly of a chromatin structure that modulates origin activity by sterically impeding Cdc45p loading. Notably, MCM proteins are not similarly excluded from late origins in G1 phase. Thus, the simple model that late origins are prevented from firing early in S by chromatin exclusion of most or all replication factors cannot accommodate these findings. Instead, there must be more subtle differences at late origins that allow limited replication complex assembly. Because MCM proteins bind to early origins slightly before Cdc45p, it is possible that MCM binding occurs before the assembly of a chromatin structure that occludes Cdc45p access. Alternatively, limiting Cdc45p dosage may restrict its association to certain origins that have greater affinity for Cdc45p. If this is the case, then overexpression of Cdc45p might enable its loading onto late origins and possibly even advance the timing of late origin activation. However, we observe no difference in the timing of Pol α or Cdc45p-origin associations when Cdc45p is overexpressed, suggesting that replication timing remains unaltered (data not shown).
How Does Cdc45p Recruit DNA Polα and Polɛ to Origins?
Inactivation of Cdc45p blocks polymerase loading and origin activation. Cdc45p may physically interact with DNA Polα to escort and load the polymerase onto the RC. This view is supported by a recent report demonstrating a requirement of Cdc45p for chromatin association of Polα and coimmunoprecipitation of Cdc45p and DNA Polα from Xenopus egg extracts (36). We were unable to coprecipitate yeast Cdc45p and DNA Polα or Polɛ from G1 or S phase extracts with or without in vivo formaldehyde-crosslinking. Alternatively, Cdc45p may stimulate a step in initiation (e.g., origin unwinding) that subsequently leads to polymerase loading. Origin DNA unwinding in SV40 replication initiation is associated with binding of replication protein A (RPA) to the single-stranded DNA (37). In yeast, origin association of RPA occurs at the G1/S transition and is independent of Polα function (38). The association of Cdc45p with the replicative complex combined with data indicating physical interaction of Cdc45p with MCM proteins (39) and DNA Polα suggests Cdc45p might function to coordinate the activities of MCM proteins and DNA polymerases at replication forks. Such a mechanism could be analogous to the function of the τ subunit of the Escherichia coli Pol III holoenzyme, which links the core DNA polymerase and the DnaB replicative helicase (40).
Regulation of Cdc45p-Origin Binding by Rad53p.
The Rad53p protein kinase is required to inhibit the activation of late-firing origins when cells initiate S phase in the presence of hydroxyurea (an inhibitor of elongation) or MMS (a DNA damaging agent). We have demonstrated that the associations of Cdc45p and DNA Polα with late origins are blocked in a Rad53p-dependent manner, and RPA-late origin association is similarly inhibited (38). As with late origin initiation, regulation of Cdc45p-origin binding provides a mechanism to inhibit origin activation by blocking the recruitment of DNA polymerase α. Although the relationship of Cdc45p to RPA in the initiation process has not been determined, the association of Cdc45p with early origins in G1 phase occurs before RPA association is detected (G1/S), suggesting Cdc45p functions upstream of RPA (Figs. 1, 2, and 3; ref. 38). If this assumption is correct, inhibition of RPA association with late origins is likely the result of the block to Cdc45p origin association. It is not known whether Rad53p directly regulates Cdc45p association to prevent initiation or whether an earlier event required for Cdc45p association is under Rad53p control. Rad53p does not inhibit MCM protein associations with late origins; however, it remains possible that Rad53p modifies MCM proteins or other pre-RC components to block Cdc45p late-origin association. Furthermore, our data do not exclude the possibility that Rad53p targets additional replication factors to block late origin activation. In a recent report, mutation of RAD53 advanced the timing of late origin activation in synchronized but otherwise unperturbed cells (30). Combined with our results, this suggests that Rad53p may function in cycling cells to control initiation timing by regulating association of Cdc45p with late origins of replication.
In conclusion, we have characterized distinct protein assemblies at early versus late origins of replication during G1 and S, providing a tool for greater understanding of replication initiation in the context of different chromosomal environments. Our results indicate that assembly of replication initiation complexes at late origins is blocked before the Cdc45p loading step and that Cdc45p is a strong candidate for a regulatory target of factors that control replication timing. Cdc45p also may mediate the Rad53p-dependent checkpoint inhibition of late origin firing, perhaps integrating S phase progression with oversight of replication fidelity.
Supplementary Material
Acknowledgments
We thank A. Amon, T. Baker, and S. Sanders for the critical reading of the manuscript and B. Stillman, P. Sorger, T. Weinert, and M. Weinreich for plasmids and strains. This work was supported in part by Searle/Chicago Community Trust and Rita Allen Foundation Awards to S.P.B. and by a grant from the National Institutes of Health (Grant RO1 GM52339). Postdoctoral Fellowships from the American Cancer Society and the Merck/M.I.T. Collaboration supported O.M.A.
ABBREVIATIONS
- ARS
autonomously replicating sequence
- RC
replicative complex
- pre-RC
pre-replicative complex
- FACS
fluorescence-activated cell sorting
- MCM
minichromosome maintenance
- CDK
cyclin-dependent kinase
- Polα
DNA polymerase α
- Polɛ
DNA polymerase ɛ
- RPA
replication protein A
References
- 1.Jacob F, Brenner S, Cuzin F. Cold Spring Harbor Symp Quant Biol. 1963;28:329–348. [Google Scholar]
- 2.DePamphilis M L. In: DNA Replication in Eukaryotic Cells. DePamphilis M L, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 45–86. [Google Scholar]
- 3.Diffley J F X. Genes Dev. 1996;10:2819–2830. doi: 10.1101/gad.10.22.2819. [DOI] [PubMed] [Google Scholar]
- 4.Diller J D, Raghuraman M K. Trends Biochem Sci. 1994;19:320–325. doi: 10.1016/0968-0004(94)90070-1. [DOI] [PubMed] [Google Scholar]
- 5.Simon I, Cedar H. In: DNA Replication in Eukaryotic Cells. DePamphilis M L, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 387–408. [Google Scholar]
- 6.Grunstein M. Cell. 1998;93:325–328. doi: 10.1016/s0092-8674(00)81160-5. [DOI] [PubMed] [Google Scholar]
- 7.Wakimoto B T. Cell. 1998;93:321–324. doi: 10.1016/s0092-8674(00)81159-9. [DOI] [PubMed] [Google Scholar]
- 8.Hsiao C L, Carbon J. Proc Natl Acad Sci USA. 1979;76:3829–3833. doi: 10.1073/pnas.76.8.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stinchcomb D T, Struhl K, Davis R W. Nature (London) 1979;282:39–43. doi: 10.1038/282039a0. [DOI] [PubMed] [Google Scholar]
- 10.Ferguson B M, Brewer B J, Reynolds A E, Fangman W L. Cell. 1991;65:507–515. doi: 10.1016/0092-8674(91)90468-e. [DOI] [PubMed] [Google Scholar]
- 11.Ferguson B M, Fangman W L. Cell. 1992;68:333–339. doi: 10.1016/0092-8674(92)90474-q. [DOI] [PubMed] [Google Scholar]
- 12.Friedman K L, Diller J D, Ferguson B M, Nyland S V, Brewer B J, Fangman W L. Genes Dev. 1996;10:1595–1607. doi: 10.1101/gad.10.13.1595. [DOI] [PubMed] [Google Scholar]
- 13.Raghuraman M K, Brewer B J, Fangman W L. Science. 1997;276:806–809. doi: 10.1126/science.276.5313.806. [DOI] [PubMed] [Google Scholar]
- 14.Newlon C S. Cell. 1997;91:717–720. doi: 10.1016/s0092-8674(00)80459-6. [DOI] [PubMed] [Google Scholar]
- 15.Diffley J F, Cocker J H, Dowell S J, Rowley A. Cell. 1994;78:303–316. doi: 10.1016/0092-8674(94)90299-2. [DOI] [PubMed] [Google Scholar]
- 16.Aparicio O M, Weinstein D M, Bell S P. Cell. 1997;91:59–69. doi: 10.1016/s0092-8674(01)80009-x. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka T, Knapp D, Nasmyth K. Cell. 1997;90:649–660. doi: 10.1016/s0092-8674(00)80526-7. [DOI] [PubMed] [Google Scholar]
- 18.Nasmyth K. Science. 1996;274:1643–1645. doi: 10.1126/science.274.5293.1643. [DOI] [PubMed] [Google Scholar]
- 19.Zou L, Stillman B. Science. 1998;280:593–596. doi: 10.1126/science.280.5363.593. [DOI] [PubMed] [Google Scholar]
- 20.Sikorski R S, Hieter P. Genetics. 1989;122:19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell S P, Kobayashi R, Stillman B. Science. 1993;262:1844–1849. doi: 10.1126/science.8266072. [DOI] [PubMed] [Google Scholar]
- 22.Strahl-Bolsinger S, Hecht A, Luo K, Grunstein M. Genes Dev. 1997;11:83–93. doi: 10.1101/gad.11.1.83. [DOI] [PubMed] [Google Scholar]
- 23.Hecht A, Strahl-Bolsinger S, Grunstein M. Nature (London) 1996;383:92–96. doi: 10.1038/383092a0. [DOI] [PubMed] [Google Scholar]
- 24.Sugino A. Trends Biochem Sci. 1995;20:319–323. doi: 10.1016/s0968-0004(00)89059-3. [DOI] [PubMed] [Google Scholar]
- 25.Dutta A, Bell S P. Annu Rev Cell Dev Biol. 1999;13:293–332. doi: 10.1146/annurev.cellbio.13.1.293. [DOI] [PubMed] [Google Scholar]
- 26.Owens J C, Detweiler C S, Li J J. Proc Natl Acad Sci USA. 1997;94:12521–12526. doi: 10.1073/pnas.94.23.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hennessy K M, Lee A, Chen E, Botstein D. Genes Dev. 1991;5:958–969. doi: 10.1101/gad.5.6.958. [DOI] [PubMed] [Google Scholar]
- 28.Weinert T. Cell. 1998;94:555–558. doi: 10.1016/s0092-8674(00)81597-4. [DOI] [PubMed] [Google Scholar]
- 29.Santocanale C, Diffley J F. Nature (London) 1998;395:615–618. doi: 10.1038/27001. [DOI] [PubMed] [Google Scholar]
- 30.Shirahige K, Hori Y, Shiraishi K, Yamashita M, Takahashi K, Obuse C, Tsurimoto T, Yoshikawa H. Nature (London) 1998;395:618–621. doi: 10.1038/27007. [DOI] [PubMed] [Google Scholar]
- 31.Diffley J F. Curr Biol. 1998;8:R771–R773. doi: 10.1016/s0960-9822(07)00483-6. [DOI] [PubMed] [Google Scholar]
- 32.Donaldson A D, Raghuraman M K, Friedman K L, Cross F R, Brewer B J, Fangman W L. Mol Cell. 1998;2:173–182. doi: 10.1016/s1097-2765(00)80127-6. [DOI] [PubMed] [Google Scholar]
- 33.Hereford L M, Hartwell L H. J Mol Biol. 1974;84:445–461. doi: 10.1016/0022-2836(74)90451-3. [DOI] [PubMed] [Google Scholar]
- 34.Donaldson A D, Fangman W L, Brewer B J. Genes Dev. 1998;12:491–501. doi: 10.1101/gad.12.4.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bousset K, Diffley J F. Genes Dev. 1998;12:480–490. doi: 10.1101/gad.12.4.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mimura S, Takisawa H. EMBO J. 1998;17:5699–5707. doi: 10.1093/emboj/17.19.5699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hubscher U, Maga G, Podust V N. In: DNA Replication in Eukaryotic Cells. DePamphilis M L, editor. Plainview, NY: Cold Spring Harbor Lab. Press; 1996. pp. 525–544. [Google Scholar]
- 38.Tanaka T, Nasmyth K. EMBO J. 1998;17:5182–5191. doi: 10.1093/emboj/17.17.5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hopwood B, Dalton S. Proc Natl Acad Sci USA. 1996;93:12309–12314. doi: 10.1073/pnas.93.22.12309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker T A, Bell S P. Cell. 1998;92:295–305. doi: 10.1016/s0092-8674(00)80923-x. [DOI] [PubMed] [Google Scholar]
- 41.Reynolds A E, McCarrol R M, Newlon C S, Fangman W L. Mol Cell Biol. 1989;9:4488–4494. doi: 10.1128/mcb.9.10.4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Friedman K L, Brewer B J, Fangman W L. Genes Cells. 1997;2:667–678. doi: 10.1046/j.1365-2443.1997.1520350.x. [DOI] [PubMed] [Google Scholar]
- 43.Yamashita M, Hori Y, Shinomiya T, Obuse C, Tsurimoto T, Yoshikawa H, Shirahige K. Genes Cells. 1997;2:655–665. doi: 10.1046/j.1365-2443.1997.1530351.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}