

Abstract
Background and aims: A low dietary folate intake can cause genomic DNA hypomethylation and may increase the risk of colorectal neoplasia. The hypothesis that folic acid supplementation increases DNA methylation in leucocytes and colorectal mucosa was tested in 31 patients with histologically confirmed colorectal adenoma using a randomised, double blind, placebo controlled, parallel design.
Methods: Subjects were randomised to receive either 400 μg/day folic acid supplement (n = 15) or placebo (n = 16) for 10 weeks. Genomic DNA methylation, serum and erythrocyte folate, and plasma homocysteine concentrations were measured at baseline and post intervention.
Results: Folic acid supplementation increased serum and erythrocyte folate concentrations by 81% (95% confidence interval (CI) 57–104%; p<0.001 v placebo) and 57% (95% CI 40–74%; p<0.001 v placebo), respectively, and decreased plasma homocysteine concentration by 12% (95% CI 4–20%; p = 0.01 v placebo). Folic acid supplementation resulted in increases in DNA methylation of 31% (95% CI 16–47%; p = 0.05 v placebo) in leucocytes and 25% (95% CI 11–39%; p = 0.09 v placebo) in colonic mucosa.
Conclusions: These results suggest that DNA hypomethylation can be reversed by physiological intakes of folic acid.
Keywords: folic acid, DNA methylation, colorectal adenoma, intervention study
Low folate intake1–3 and low serum4,5 and erythrocyte4,6,7 folate concentrations may increase the risk of colorectal adenoma and cancer. An intervention study using adenoma recurrence as an end point reported a 46% decrease in adenoma recurrence in 31 subjects receiving 1 mg/day folate for two years after polypectomy compared with 29 subjects receiving placebo.8
Low folate status may increase the risk of neoplasia by inducing DNA hypomethylation, which can affect DNA stability and gene expression. Folate is a methyl donor in the methylation cycle, which maintains adequate cellular levels of S-adenosylmethionine (SAM) for biological methylation reactions, including methylation of DNA. The byproduct of this reaction is homocysteine, which can either be irreversibly removed by the enzyme cystathionine-β-synthase or reconverted back to methionine and then SAM in a reaction catalysed by the vitamin B12 dependent enzyme methionine synthase (MS). The methyl group for this reaction is provided by 5-methyltetrahydrofolate, supplied via the enzyme 5,10 methylenetetrahydrofolate reductase (MTHFR). A limited supply of folate can impair the methylation cycle, and consequently DNA methylation, and cause intracellular levels of homocysteine to rise.
DNA hypomethylation has been reported in colorectal tumour tissue9,10 and normal appearing colorectal mucosa of patients with carcinomas.11 Hypomethylation of DNA from normal appearing colorectal mucosa has been associated with lower folate status and risk of colorectal adenoma and cancer.12 Genomic DNA hypomethylation is accompanied by altered methylation in the promoter regions of critical genes. Promoter hypermethylation generally suppresses transcription leading to inactivation of tumour suppressor genes,13–15 while promoter hypomethylation can activate proto-oncogenes16 and has also been associated with loss of imprinting,17 which occurs commonly in both tumour and normal tissue of patients with colorectal cancer.
Two polymorphisms in genes coding for enzymes involved in the methylation cycle, methylenetetrahydrofolate reductase, MTHFR 677C→T and methionine synthase, MS 2756A→G, have been associated with a reduced risk of colorectal cancer18–25 but not adenoma.26–28 However, the apparent protective effects on colorectal cancer disappear under conditions of low folate status and/or high alcohol intake. The MTHFR 677C→T mutation in combination with low folate/high alcohol intake has been shown to increase adenoma risk.26,28 The MTHFR 677C→T26,28 and MS 2756A→G30 mutations have been associated with increased and decreased plasma homocysteine (a biomarker of the methylation cycle) concentrations, respectively. Studies have also reported associations between the MTHFR 677C→T polymorphism and variation in genomic methylation levels in leucocytes from healthy individuals31,32 although it is not clear what impact these polymorphisms have on DNA methylation in colorectal mucosa. A recent folate depletion–repletion study showed a greater increase in DNA methylation in leucocytes following repletion in young women homozygous for the MTHFR 677C→T polymorphism compared with those who did not carry the mutation.33 Recent studies suggest that the MTHFR 677C→T polymorphism may be linked to promoter hypermethylation and lower concentrations of folate intermediates in colorectal tumours.34,35
Previous studies in subjects with colorectal neoplasia using genomic DNA methylation in colorectal mucosa as a primary end point suggest that folate supplementation can reverse DNA hypomethylation.11,36,37 However, these studies had relatively small sample sizes (11–20 subjects) and used 1–10 mg of folic acid daily, which is 5–50 times greater than the UK reference nutrient intake (RNI) and 2.5–25 times greater than the US recommended dietary allowance (RDA) for folate. There are concerns that high intakes of folic acid can mask vitamin B12 deficiency, resulting in the development of irreversible neurological symptoms. The aim of the present study therefore was to test the hypothesis that a physiological dose of folic acid would increase DNA methylation in patients with colorectal adenoma. We used individuals with adenoma as they are at increased risk of developing colorectal cancer but are free of the possible confounding effects that malignant disease may have on folate status.
MATERIALS AND METHODS
Patients
Subjects were recruited serially from all patients attending for a clinically indicated colonoscopy at the Department of Colorectal Surgery, King’s College Hospital, London, between August 2000 and March 2001. All patients recruited into the intervention trial were participants in a previously reported case control study.12 Details of patient recruitment and preparation for colonoscopy are described elsewhere.12 Of 196 patients screened, 32 met the inclusion criteria and consented to participate in the trial. Inclusion criteria were histologically confirmed colorectal adenoma at current colonoscopy. Exclusion criteria were current or previous diagnosis of colorectal cancer, a strong family history of colorectal cancer or adenomatous polyposis coli, inflammatory bowel disease, a current or past history of gluten sensitive enteropathy, clinical and/or laboratory evidence of intestinal malabsorption, significant renal or liver disease, pregnancy, epilepsy, alcoholism, pernicious anaemia, serum vitamin B12 levels <180 ng/l, and the use of antifolate medication or dietary supplements containing folic acid and/or vitamin B12.
Weight and height were recorded and patients provided information on smoking habits, current medication, and supplement use, and completed a specially designed food frequency questionnaire (FFQ) to assess dietary folate and alcohol intakes.38 Patients were asked to bring in any medication and/or supplement containers to check that they were suitable for inclusion in the intervention study. Written informed consent was obtained from all patients according to the guidelines of the Research Ethics Committee at King’s College Hospital NHS Trust and King’s College London.
Sample size, randomisation, and treatment allocation
Patients were randomised to receive either 400 μg folic acid or an identical appearing placebo (both gifts from RP Scherer, UK) for 10 weeks. Sample size was calculated to detect 1 SD change in DNA methylation in colorectal mucosa with 80% power and an α error of 0.05, which indicated 16 patients in each group. Randomisation was generated by a computer in blocks of six and stratified by sex. Numbered containers were used to implement the random allocation sequence, which was concealed until interventions were assigned. Treatment allocation was concealed from patients and all researchers involved in administering the interventions and assessing the outcomes. Subjects were advised not to alter their diet or usual lifestyle in any way for the entire duration of the trial.
Blood and tissue sampling
Fasting venous blood samples were obtained at baseline and after the intervention for determination of serum and erythrocyte folate concentrations, plasma homocysteine, genomic DNA methylation, and MTHFR 677C→T and MS 2756A→G polymorphisms. Baseline blood samples were obtained on the day of colonoscopy prior to the procedure. Rectal biopsies of normal appearing mucosa for determination of genomic DNA methylation were obtained during colonoscopy (baseline) and by rigid sigmoidoscopy after the treatment. On both occasions mucosal biopsy specimens were removed from the rectum (about 12 cm from the anal verge) at least 5 cm from any lesion or mucosal abnormality and immediately snap frozen in liquid nitrogen. All patients had prepared for the colonoscopy by taking an orally administered colonic lavage solution (KleanPrep; Norgine, Harefield, UK). For the rigid sigmoidoscopy, patients were instructed to empty their bowels using two suppositories on the morning of their appointment. No abnormalities were detected in any of the subjects on sigmoidoscopy. Patients were asked to return their remaining supplement pills for counting to check compliance.
The laboratory methods used to analyse serum and erythrocyte folate concentrations, plasma homocysteine, genomic DNA methylation, and gene polymorphisms have been described in detail previously.12 Genomic DNA methylation was determined in leucocytes and colonic mucosa using an in vitro [3H] methyl acceptance assay.39 In this assay a decrease in methyl group incorporation in DNA indicates an increase in methylation status.
Study end points and statistical analysis
The primary end points were changes in DNA methylation in colorectal mucosa and leucocytes. Secondary end points were changes in serum and erythrocyte folate and plasma homocysteine concentrations. All patients were analysed on an intention to treat basis. Analyses were performed using Intercooled Stata (version 6.0 for Windows, Stata Corporation, College Station, Texas, USA). χ2 tests and analysis of variance were used to compare the folic acid and placebo groups for differences in categorical variables (alcohol intake, smoking, genotype, and clinical characteristics) and continuous variables (folate intake and serum vitamin B12 concentrations), respectively. A Mann-Whitney U test was used to compare the folic acid and placebo groups for differences in body mass index as this variable was not normally distributed.
All continuous variables for the primary and secondary end points, except plasma homocysteine, were normally distributed. For plasma homocysteine, logarithmic transformation did not entirely remove the positive skewness. However, exclusion of an outlier (plasma homocysteine >30 μmol/l) normalised the data and therefore analyses for homocysteine were performed after excluding this patient. Analysis of variance was used to obtain adjusted means for baseline and post intervention values of serum and erythrocyte folate, plasma homocysteine, and [3H] methyl incorporation in colorectal mucosa and leucocytes for the folic acid and placebo groups, after adjusting for sex, age, body mass index, smoking, alcohol intake, and genotype. Paired t tests were used to assess differences between baseline and post intervention values in each group. The relationships between continuous variables were examined using Pearson correlation coefficients.
In order to compare changes between the two time points in the folic acid and placebo groups, baseline values were subtracted from post intervention values for each subject for all variables measured (serum and erythrocyte folate, plasma homocysteine, and [3H] methyl incorporation in colorectal mucosa and leucocytes). Analysis of variance was then used to compare mean changes in these variables between the folic acid and placebo groups, with adjustment for sex, age, body mass index, smoking, alcohol intake, and genotype. Statistical significance was set at the 5% level.
RESULTS
Of the 32 patients enrolled who met the inclusion criteria, 31 patients completed the study; one patient withdrew because of another comorbidity (fig 1 ▶). Baseline and clinical characteristics of the subjects are shown in tables 1 ▶ and 2 ▶, respectively.
Figure 1.
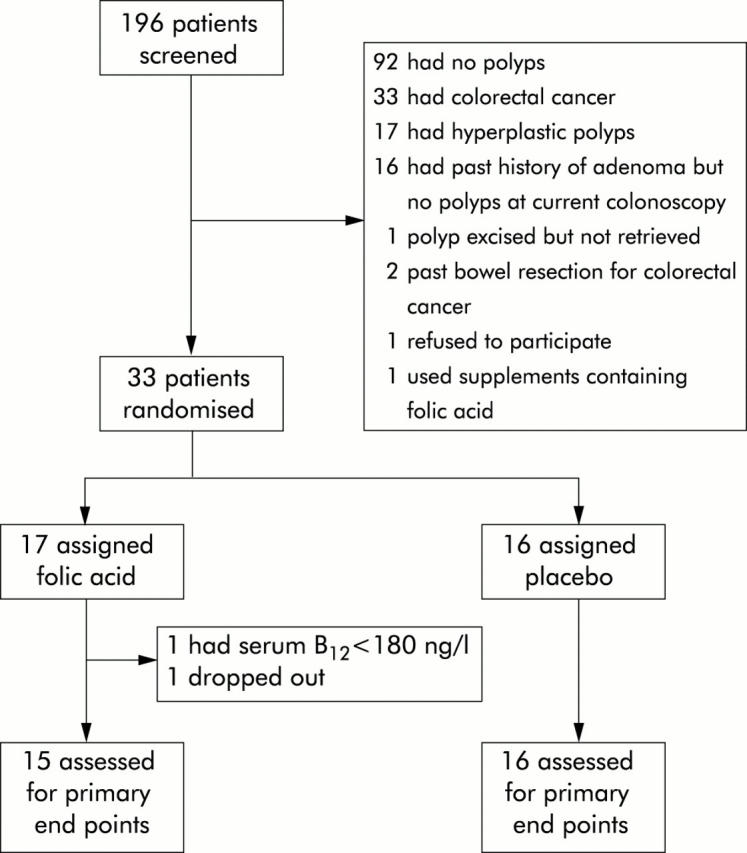
Trial profile.
Table 1.
Baseline characteristics of the patients by treatment group
| Folic acid (n = 15) | Placebo (n = 16) | ||
| n (M/F) | 7/8 | 8/8 | |
| Age (y) | 63.9 (11.9) | 63.8 (9.7) | |
| Body mass index (kg/m2) | 25.3 (19.8–49.4) | 25.0 (19.8–36.8) | |
| Folate intake (μg/day) | 341 (118) | 368 (156)† | |
| Serum vitamin B12 (ng/l) | 446 (235) | 461 (235) | |
| Alcohol intake (g/day) | |||
| Lower third (<2) | 7 (47) | 3 (19) | |
| Middle third (2–37) | 5 (33) | 6 (38) | |
| Upper third (>37) | 3 (20) | 7 (44) | |
| Smoking | |||
| Current smoker | 1 (7) | 8 (50)** | |
| Non-smoker | 14 (93) | 8 (50) | |
| MTHFR 677C→T genotype | |||
| CC | 10 (67) | 9 (56) | |
| CT | 5 (33) | 5 (31) | |
| TT | 0 (0) | 2 (13) | |
| MS 2756A→G genotype | |||
| AA | 10 (67) | 6 (38) | |
| AG | 5 (33) | 9 (56) | |
| GG | 0 (0) | 1 (6) | |
MTHFR, methylenetetrahydrofolate reductase; MS, methionine synthase.
Values are arithmetic means (SD) for age, folate intake, and serum vitamin B12, median (range) for body mass index, and number (%) for alcohol intake, smoking, and genotype.
**p<0.01 compared with the folic acid group.
†n = 15.
Table 2.
Clinical characteristics of the patients by treatment group
| Folic acid (n = 15) | Placebo (n = 16) | |
| No of adenomas | ||
| 1 | 11 (73%) | 11 (69%) |
| 1–3 | 3 (20%) | 5 (31%) |
| >3 | 1 (7%) | 0 (0%) |
| Type of adenoma | ||
| Tubular | 2 (13%) | 1 (6%) |
| Tubulovillous | 11 (73%) | 12 (75%) |
| Villous | 2 (13%) | 2 (13%) |
| Serrated | 0 (0%) | 1 (6%) |
| Degree of dysplasia | ||
| Mild | 3 (20%) | 2 (13%) |
| Moderate | 9 (60%) | 9 (56%) |
| Severe | 3 (20%) | 4 (25%) |
Values are number (%) of patients.
There was a greater proportion of smokers in the placebo group (50%) compared with the folic acid group (7%) but there were no other significant differences in baseline or clinical characteristics between the two groups. All subsequent analyses were performed after adjusting for smoking, as well as for sex, age, body mass index, alcohol intake, and genotype. Measurements of serum and erythrocyte folate were positively correlated at baseline in all subjects (r = 0.661, p<0.001). Baseline plasma homocysteine concentrations correlated negatively with both serum (r = −0.544, p<0.01) and erythrocyte folate (r = −0.611, p<0.001). Folate intake, as assessed by the FFQ, was correlated positively with serum (r = 0.385, p = 0.04) and erythrocyte folate (r = 0.525, p = 0.03) and negatively with plasma homocysteine (r = −0.376, p = 0.04).
Table 3 ▶ shows serum and erythrocyte folate, plasma homocysteine, and markers of DNA methylation before and after supplementation with folic acid or placebo. There were no significant differences between the folic acid and placebo groups at baseline. Compared with baseline, folate supplementation increased serum and erythrocyte folate concentrations by 81% (95% confidence interval (CI) 57–104%; p<0.001) and 57% (95% CI 40–74%; p<0.001) respectively, decreased plasma homocysteine concentration by 12% (95% CI 4–20%; p = 0.04), and increased DNA methylation by 31% in leucocytes (95% CI 16–47%; p = 0.001) and by 25% in colonic mucosa (95% CI 11–39%; p = 0.02). In the placebo group, there were no significant changes from baseline. Mean changes were significantly different between the folic acid and placebo groups for serum and erythrocyte folate concentrations (both p<0.001) and plasma homocysteine (p = 0.01), and of borderline significance for DNA methylation in leucocytes (p = 0.05) and in colonic mucosa (p = 0.09). For plasma homocysteine, inclusion of the outlier with plasma homocysteine concentrations >30 mmol/l increased the significance of the results.
Table 3.
Serum and erythrocyte folate, plasma homocysteine, and markers of DNA methylation before and after supplementation with folic acid or placebo
| Folic acid (n = 15) | Placebo (n = 16) | p Value | |
| Serum folate* (μg/l) | |||
| Baseline | 7.4 (5.7 to 9.2)§ | 8.2 (6.6 to 9.8) | |
| Follow up | 13.4 (11.6 to 15.2)§ | 7.3 (5.6 to 8.9) | |
| Change | +6.0 (4.2 to 7.7)§ | −1.0 (−2.6 to 0.7) | <0.001 |
| Erythrocyte folate* (μg/l) | |||
| Baseline | 282 (227 to 387) | 316 (261 to 371)¶ | |
| Follow up | 443 (376 to 511) | 305 (238 to 373)¶ | |
| Change | +161 (114 to 209) | −11 (−58 to 7)¶ | <0.001 |
| Plasma homocysteine*† (μmol/l) | |||
| Baseline | 12.2 (10.1 to 14.4)§ | 11.5 (9.5 to 13.5) | |
| Follow up | 10.7 (8.4 to 13.1)§ | 12.3 (10.2 to 14.5) | |
| Change | −1.5 (−2.5 to −0.5)§ | +0.8 (−0.1 to 1.8) | 0.01 |
| [3H] methyl incorporation in leucocyte DNA*† (Bq/μg DNA) | |||
| Baseline | 748 (672 to 825) | 678 (605 to 752) | |
| Follow up | 515 (440 to 589) | 655 (583 to 727) | |
| Change | −233 (−349 to −118) | −23 (−134 to 88) | 0.05 |
| [3H] methyl incorporation in colonic DNA*† (Bq/μg DNA) | |||
| Baseline | 602 (515 to 689) | 473 (389 to 556) | |
| Follow up | 451 (361 to 541) | 458 (371 to 544) | |
| Change | −151 (−234 to −69) | −15 (−95 to 64) | 0.09 |
MTHFR, methylenetetrahydrofolate reductase; MS, methionine synthase.
Values are arithmetic means (95% confidence interval) adjusted for sex, age, body mass index, smoking, alcohol intake, *MTHFR 677C→T genotype, and †MS 2756A→G genotype.
§n = 14.
¶n = 15.
DISCUSSION
The results of this trial show that daily supplementation with 400 μg/day folic acid for 10 weeks resulted in a significant fall in plasma homocysteine concentrations and a marginal increase in leucocyte DNA methylation. There was also an increase in rectal mucosa DNA methylation although this increase was not significantly different from that seen in the placebo group. The magnitude of this change was equivalent to 0.83 standard deviations (95% CI 0.10–1.57), which is less than that assumed in the original power calculation. The revised power calculation on the basis of our results suggests 23 patients per group. We made adjustments for polymorphisms in genes coding for enzymes involved in the methylation cycle, which have been shown to influence DNA methylation, as well as factors such as age and alcohol intake, which may also influence DNA methylation. DNA methylation was determined by measuring [3H] methyl incorporation into DNA and therefore a decrease in [3H] methyl incorporation reflects an increase in DNA methylation status.
Three previous randomised placebo controlled folate intervention trials showed an increase in DNA methylation in colonic mucosa in subjects with adenoma following intakes of folic acid that were 2.5–25 times those used in the present study.11,36,37 Supplementation with 10 mg/day folate for six months increased DNA methylation in colonic mucosa in seven patients with resected adenoma and four patients with cancer.11 In a trial with 20 subjects with adenoma receiving 5 mg/day folate or placebo for three months, followed by cross over to the alternative treatment for another three months, folate supplementation increased DNA methylation in colonic mucosa in those with a single polyp.36 Similarly, a study in which 20 patients with adenoma were randomised to receive either 5 mg/day folate (n = 10) or placebo (n = 10) for one year after polypectomy found a significant increase in DNA methylation in colonic mucosa at six months and one year in the folate group.37 In this study, there was also a significant increase in colonic DNA methylation in the placebo group at one year, despite the fact that blood folate and colonic folate concentrations were significantly lower than in subjects in the folic acid group at both time points.
Although the statistical effect was marginal, the present study is the first to report an increase in DNA methylation in subjects with adenoma using a physiological dose of folate for a relatively short intervention period. It is also the first to report an increase in leucocyte DNA methylation following folate supplementation in subjects with adenoma. DNA methylation in leucocytes has been shown to decrease in response to moderate folate depletion in healthy postmenopausal40,41 and young33 women. Furthermore, a study in healthy young women showed that mild elevations in plasma homocysteine were associated with a parallel increase in plasma S-adenosyl homocysteine, which correlated well with leucocyte DNA hypomethylation (r = 0.74, p<0.001).42 However, it is not clear how quickly DNA methylation responds to folate repletion or the optimal dose required to reverse hypomethylation. In the present study, the patients’ mean dietary folate intakes were below the US RDA of 400 μg/day dietary folate equivalents but above the RNI for folate in the UK (200 μg/day). Plasma and erythrocyte folate concentrations were within laboratory normal ranges (>3 μg/l and >150 μg/l for serum and erythrocyte folate, respectively) although plasma homocysteine concentrations were mildly elevated compared with population averages for this age group.43,44 A study in healthy young women reported that leucocyte DNA methylation was significantly lower in women with homocysteine concentrations of 9.3–16.5 μmol/l than in those who had homocysteine concentrations of 5.8–8.7 μmol/l.42
In colorectal neoplasia, genomic DNA hypomethylation is accompanied by region specific hyper/hypomethylation in critical genes. There is evidence that these events are not confined solely to the neoplasm but also occur in normal appearing colorectal mucosa.15,45 A recent study investigating the effects of folate and alcohol intake on promoter methylation in sporadic colorectal cancer suggested that the prevalence of hypermethylation was higher in cancers derived from patients with low folate/high alcohol intake than in cancers from patients with high folate/low alcohol intake.46 Furthermore, in a recent placebo controlled intervention trial, supplementation with 5 mg/day folic acid prevented loss of heterozygosity in the DCC tumour suppressor gene.47 Loss of heterozygosity is implicated in the initiation and progression of many cancers and has been linked to aberrant DNA methylation in promoter regions of genes. Age related hypermethylation of CpG rich promoter regions in the oestrogen receptor (ER) and mismatch repair (MLH1) genes in normal colonic mucosa has been reported.48,49 It is not clear what causes these changes but the possible link between dietary factors such as folate and age related hypermethylation in critical genes should be investigated in future studies. It is possible that changes in DNA methylation levels following folic acid supplementation may have been greater in normal mucosa from the proximal colon compared with those observed in the rectal mucosa as colorectal tumours with aberrant DNA methylation arise predominantly in the proximal colon.50
Patients with hyperplastic polyps were excluded from the present study because at the time the study was conducted there was limited evidence for the role of hyperplastic polyps in colorectal cancer. However, recent evidence has implicated hyperplastic polyps in the pathogenesis of a type of colorectal cancer characterised by extensive region specific DNA hypermethylation and microsatellite instability.51 The only study to investigate global DNA methylation in hyperplastic polyps has shown that the extent of hypomethylation in hyperplastic polyps was similar to that observed in adenomas.52 Hence future studies assessing the role of dietary factors on DNA methylation and the development of neoplasia should also include patients with hyperplastic polyps.
In conclusion, our results support the view that suboptimal intakes of folate may lead to hypomethylation, which in turn could be involved in the aetiology of colorectal cancer. Larger studies are required to confirm these findings and to assess the effects of reversing genomic DNA hypomethylation on colorectal cancer risk. However, the impact of an increased supply of folic acid in an at risk population would need to be carefully evaluated as folate may promote tumour growth in individuals with unconfirmed colorectal neoplasms, as has been demonstrated in animal studies.53,54
Abbreviations
MTHFR, methylenetetrahydrofolate reductase
MS, methionine synthase
SAM, S-adenosylmethionine
RDA, recommended dietary allowance
RNI, reference nutrient intake
FFQ, food frequency questionnaire
Conflict of interest: None declared.
REFERENCES
- 1.Giovannucci E, Stampfer MJ, Colditz GA, et al. Folate, methionine, and alcohol intake and risk of colorectal adenoma. J Natl Cancer Inst 1993;85:875–84. [DOI] [PubMed] [Google Scholar]
- 2.Giovannucci E, Rimm EB, Ascherio A, et al. Alcohol, low-methionine-low folate diets, and risk of colon cancer in men. J Natl Cancer Inst 1995;87:265–73. [DOI] [PubMed] [Google Scholar]
- 3.Giovannucci E, Stampfer MJ, Colditz GA, et al. Multivitamin use, folate, and colon cancer in women in the Nurses’ Health Study. Ann Intern Med 1998;129:517–24. [DOI] [PubMed] [Google Scholar]
- 4.Bird CL, Swendseid ME, Witte JS, et al. Red cell and plasma folate, folate consumption, and the risk of colorectal adenomatous polyps. Cancer Epidemiol Biomarkers Prev 1995;4:709–14. [PubMed] [Google Scholar]
- 5.Kato I, Dnistrian AM, Schwartz M, et al. Serum folate, homocysteine and colorectal cancer risk in women: a nested case-control study. Br J Cancer 1999;79:1917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lashner BA. Red blood cell folate is associated with the development of dysplasia and cancer in ulcerative colitis. J Cancer Res Clin Oncol 1993;119:549–54. [DOI] [PubMed] [Google Scholar]
- 7.Paspatis GA, Kalafatis E, Oros L, et al. Folate status and adenomatous colonic polyps. A colonoscopically controlled study. Dis Colon Rectum 1995;38:64–7. [DOI] [PubMed] [Google Scholar]
- 8.Paspatis GA, Karamanolis DG. Folate supplementation and adenomatous colonic polyps. Dis Colon Rectum 1994;37:1340–1. [DOI] [PubMed] [Google Scholar]
- 9.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 1983;301:89–92. [DOI] [PubMed] [Google Scholar]
- 10.Goelz SE, Vogelstein B, Hamilton SR, et al. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 1985;228:187–90. [DOI] [PubMed] [Google Scholar]
- 11.Cravo M, Fidalgo P, Pereira AD, et al. DNA methylation as an intermediate biomarker in colorectal cancer: modulation by folic acid supplementation. Eur J Cancer Prev 1994;3:473–9. [DOI] [PubMed] [Google Scholar]
- 12.Pufulete M, Al Ghnaniem R, Leather AJ, et al. Folate status, genomic DNA hypomethylation, and risk of colorectal adenoma and cancer: a case control study. Gastroenterology 2003;124:1240–8. [DOI] [PubMed] [Google Scholar]
- 13.Hiltunen MO, Alhonen L, Koistinaho J, et al. Hypermethylation of the APC (adenomatous polyposis coli) gene promoter region in human colorectal carcinoma. Int J Cancer 1997;70:644–8. [DOI] [PubMed] [Google Scholar]
- 14.Iacopetta B . Aberrant DNA methylation: have we entered the era of more than one type of colorectal cancer? Am J Pathol 2003;162:1043–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Issa JP, Ottaviano YL, Celano P, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994;7:536–40. [DOI] [PubMed] [Google Scholar]
- 16.Sharrard RM, Royds JA, Rogers S, et al. Patterns of methylation of the c-myc gene in human colorectal cancer progression. Br J Cancer 1992;65:667–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cui H, Onyango P, Brandenburg S, et al. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res 2002;62:6442–6. [PubMed] [Google Scholar]
- 18.Chen J, Giovannucci E, Kelsey K, et al. A methylenetetrahydrofolate reductase polymorphism and the risk of colorectal cancer. Cancer Res 1996;56:4862–4. [PubMed] [Google Scholar]
- 19.Chen J, Ma J, Stampfer MJ, et al. Linkage disequilibrium between the 677C→T and 1298A→C polymorphisms in human methylenetetrahydrofolate reductase gene and their contributions to risk of colorectal cancer. Pharmacogenetics 2002;12:339–42. [DOI] [PubMed] [Google Scholar]
- 20.Keku T, Millikan R, Worley K, et al. 5,10-Methylenetetrahydrofolate reductase codon 677 and 1298 polymorphisms and colon cancer in African Americans and whites. Cancer Epidemiol Biomarkers Prev 2002;11:1611–21. [PubMed] [Google Scholar]
- 21.Ma J, Stampfer MJ, Giovannucci E, et al. Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res 1997;57:1098–102. [PubMed] [Google Scholar]
- 22.Shannon B, Gnanasampanthan S, Beilby J, et al. A polymorphism in the methylenetetrahydrofolate reductase gene predisposes to colorectal cancers with microsatellite instability. Gut 2002;50:520–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma J, Stampfer MJ, Christensen B, et al. A polymorphism of the methionine synthase gene: association with plasma folate, vitamin B12, homocyst(e)ine, and colorectal cancer risk. Cancer Epidemiol Biomarkers Prev 1999;8:825–9. [PubMed] [Google Scholar]
- 24.Matsuo K, Hamajima N, Hirai T, et al. Methionine synthase reductase gene A66G polymorphism is associated with risk of colorectal cancer. Asian Pac J Cancer Prev 2002;3:353–9. [PubMed] [Google Scholar]
- 25.Toffoli G, Gafa R, Russo A, et al. Methylenetetrahydrofolate reductase 677 C→T polymorphism and risk of proximal colon cancer in north Italy. Clin Cancer Res 2003;9:743–8. [PubMed] [Google Scholar]
- 26.Levine AJ, Siegmund KD, Ervin CM, et al. The methylenetetrahydrofolate reductase 677C→T polymorphism and distal colorectal adenoma risk. Cancer Epidemiol Biomarkers Prev 2000;9:657–63. [PubMed] [Google Scholar]
- 27.Marugame T, Tsuji E, Inoue H, et al. Methylenetetrahydrofolate reductase polymorphism and risk of colorectal adenomas. Cancer Lett 2000;151:181–6. [DOI] [PubMed] [Google Scholar]
- 28.Ulrich CM, Kampman E, Bigler J, et al. Colorectal adenomas and the C677T MTHFR polymorphism: evidence for gene-environment interaction? Cancer Epidemiol Biomarkers Prev 1999;8:659–68. [PubMed] [Google Scholar]
- 29.Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet 1995;10:111–13. [DOI] [PubMed] [Google Scholar]
- 30.Harmon DL, Shields DC, Woodside JV, et al. Methionine synthase D919G polymorphism is a significant but modest determinant of circulating homocysteine concentrations. Genet Epidemiol 1999;17:298–309. [DOI] [PubMed] [Google Scholar]
- 31.Friso S, Choi SW, Girelli D, et al. A common mutation in the 5,10-methylenetetrahydrofolate reductase gene affects genomic DNA methylation through an interaction with folate status. Proc Natl Acad Sci U S A 2002;99:5606–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stern LL, Mason JB, Selhub J, et al. Genomic DNA hypomethylation, a characteristic of most cancers, is present in peripheral leukocytes of individuals who are homozygous for the C677T polymorphism in the methylenetetrahydrofolate reductase gene. Cancer Epidemiol Biomarkers Prev 2000;9:849–53. [PubMed] [Google Scholar]
- 33.Shelnutt KP, Kauwell JP, Gregory J Fiii. et al Methylenetetrahydrofolate reductase 677C→T polymorphism affects DNA methylation in response to controlled folate intake in young women. J Nutr Biochem 2004;15:554–60. [DOI] [PubMed] [Google Scholar]
- 34.Oyama K, Kawakami K, Maeda K, et al. The association between methylenetetrahydrofolate reductase polymorphism and promoter methylation in proximal colon cancer. Anticancer Res 2004;24:649–54. [PubMed] [Google Scholar]
- 35.Kawakami K, Ruszkiewicz A, Bennett G, et al. The folate pool in colorectal cancers is associated with DNA hypermethylation and with a polymorphism in methylenetetrahydrofolate reductase. Clin Cancer Res 2003;9:5860–5. [PubMed] [Google Scholar]
- 36.Cravo ML, Pinto AG, Chaves P, et al. Effect of folate supplementation on DNA methylation of rectal mucosa in patients with colonic adenomas: correlation with nutrient intake. Clin Nutr 1998;17:45–9. [DOI] [PubMed] [Google Scholar]
- 37.Kim YI, Baik HW, Fawaz K, et al. Effects of folate supplementation on two provisional molecular markers of colon cancer: a prospective, randomized trial. Am J Gastroenterol 2001;96:184–95. [DOI] [PubMed] [Google Scholar]
- 38.Pufulete M, Emery PW, Nelson M, et al. Validation of a short food frequency questionnaire to assess folate intake. Br J Nutr 2002;87:383–90. [DOI] [PubMed] [Google Scholar]
- 39.Balaghi M, Wagner C. DNA methylation in folate deficiency: use of CpG methylase. Biochem Biophys Res Commun 1993;193:1184–90. [DOI] [PubMed] [Google Scholar]
- 40.Jacob RA, Gretz DM, Taylor PC, et al. Moderate folate depletion increases plasma homocysteine and decreases lymphocyte DNA methylation in postmenopausal women. J Nutr 1998;128:1204–12. [DOI] [PubMed] [Google Scholar]
- 41.Rampersaud GC, Kauwell GP, Hutson AD, et al. Genomic DNA methylation decreases in response to moderate folate depletion in elderly women. Am J Clin Nutr 2000;72:998–1003. [DOI] [PubMed] [Google Scholar]
- 42.Yi P, Melnyk S, Pogribna M, et al. Increase in plasma homocysteine associated with parallel increases in plasma S-adenosylhomocysteine and lymphocyte DNA hypomethylation. J Biol Chem 2000;275:29318–23. [DOI] [PubMed] [Google Scholar]
- 43.Jacques PF, Bostom AG, Wilson PW, et al. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am J Clin Nutr 2001;73:613–21. [DOI] [PubMed] [Google Scholar]
- 44.Selhub J, Jacques PF, Rosenberg IH, et al. Serum total homocysteine concentrations in the third National Health and Nutrition Examination Survey (1991–1994): population reference ranges and contribution of vitamin status to high serum concentrations. Ann Intern Med 1999;131:331–9. [DOI] [PubMed] [Google Scholar]
- 45.Ahuja N, Li Q, Mohan AL, et al. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res 1998;58:5489–94. [PubMed] [Google Scholar]
- 46.van Engeland M, Weijenberg MP, Roemen GM, et al. Effects of dietary folate and alcohol intake on promoter methylation in sporadic colorectal cancer: the Netherlands cohort study on diet and cancer. Cancer Res 2003;63:3133–7. [PubMed] [Google Scholar]
- 47.Nagothu KK, Jaszewski R, Moragoda L, et al. Folic acid mediated attenuation of loss of heterozygosity of DCC tumor suppressor gene in the colonic mucosa of patients with colorectal adenomas. Cancer Detect Prev 2003;27:297–304. [DOI] [PubMed] [Google Scholar]
- 48.Nakagawa H, Nuovo GJ, Zervos EE, et al. Age-related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res 2001;61:6991–5. [PubMed] [Google Scholar]
- 49.Issa JP, Ottaviano YL, Celano P, et al. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet 1994;7:536–40. [DOI] [PubMed] [Google Scholar]
- 50.Hawkins N, Norrie M, Cheong K, et al. CpG island methylation in sporadic colorectal cancers and its relationship to microsatellite instability. Gastroenterology 2002;122:1376–87. [DOI] [PubMed] [Google Scholar]
- 51.Jass JR. Methylation patterns define two types of hyperplastic polyp associated with colorectal cancer. Clin Gastroenterol Hepatol 2004;2:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bariol C, Suter C, Cheong K, et al. The relationship between hypomethylation and CpG island methylation in colorectal neoplasia. Am J Pathol 2003;162:1361–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Song J, Medline A, Mason JB, et al. Effects of dietary folate on intestinal tumorigenesis in the apcMin mouse. Cancer Res 2000;60:5434–40. [PubMed] [Google Scholar]
- 54.Bashir O, FitzGerald AJ, Goodlad RA. Both suboptimal and elevated vitamin intake increase intestinal neoplasia and alter crypt fission in the ApcMin/+ mouse. Carcinogenesis 2004;25:1507–15. [DOI] [PubMed] [Google Scholar]