

Abstract
Background and aims: Thrombospondin 1 (TSP-1) is an important activator of latent transforming growth factor β (TGF-β) but little is known of the expression patterns and functions of TSP-1 in liver cells. We therefore analysed if and how TSP-1 acts on TGF-β during fibrogenesis.
Methods and results: Using reverse transcription-polymerase chain reaction, we demonstrated that hepatocytes from normal liver expressed no TSP-1 mRNA whereas Kupffer cells and sinusoidal endothelial cells did. TSP-1 mRNA and protein were detected in quiescent and activated cultured hepatic stellate cells (HSC) and TSP-1 expression was highly inducible by platelet derived growth factor BB (PDGF-BB) and, to a lesser extent, by tumour necrosis factor α in activated HSC. Furthermore, addition of PDGF-BB directly led to enhanced TGF-β mRNA expression and a TSP-1 dependent increase in TGF-β/Smad signalling. Using either a peptide specifically blocking the interaction of TSP-1 with latent TGF-β or antibodies against TSP-1 not only abrogated activation of latent TGF-β but also reduced the effects of the active dimer itself.
Conclusions: Our data suggest that TSP-1 expression is important for TGF-β effects and that it is regulated by the profibrogenic mediator PDGF-BB in HSC. Furthermore, the presence of TSP-1 seems to be a prerequisite for effective signal transduction by active TGF-β not only in rat HSC but also in other cell types such as human dermal fibroblasts.
Keywords: transforming growth factor, platelet derived growth factor, tumour necrosis factor, liver, fibrosis, thrombospondin
Thrombospondins form a family of secreted glycoproteins with pleiotropic functions and widespread expression.1 They can be divided into two subgroups: (i) TSP-1 and TSP-2 are highly homologous trimers with a chain molecular mass of approximately 145 kDa and (ii) TSP-3–TSP-5 are pentamers with chain molecular masses of approximately 100 kDa having functions distinct from those of TSP-1 and TSP-2. In this study, we focused our work on regulation of TSP-1 because its presence, cellular origin, and potential functions in the liver are largely unknown. Although TSP-1 and TSP-2 share a similar domain structure, their different developmental and spatial patterns of expression during embryogenesis2,3 and the distinct phenotypes of the corresponding knockout mice4,5 indicate specific non-overlapping functions. Furthermore, their expression is mediated by distinct promoter sequences.6–8 Both TSP-1 and TSP-2 are involved in the regulation of cellular responses to injury 9,10 and for TSP-1, but not for TSP-2, an increasingly important role as activator of the small latent transforming growth factor β (TGF-β) complex11,12 has been defined. In addition, increased expression of TSP-1 has been found in livers from patients who suffer from congenital hepatic fibrosis13 and in liver specimens from patients with hepatocellular carcinoma.14 Blocking interactions of TSP-1 with latent TGF-β has recently been demonstrated to inhibit experimentally induced fibrogenesis in rats.15 These data strengthen the potential importance of TSP-1 in hepatic activation of latent TGF-β in vivo but the underlying molecular mechanisms, cell types, and mediators involved in this process are still largely unknown.
The responses of liver cells to injury, including wound healing, inflammation, and fibrosis are mediated by an array of cytokines, including, among others, TGF-β, platelet derived growth factor (PDGF), and tumour necrosis factor α (TNF-α). During these processes, hepatic stellate cells (HSC) become activated and transdifferentiate into matrix producing myofibroblasts (MFB). These fibrogenic cells are an important cellular source of latent TGF-β which is secreted as a large latent TGF-β complex16 in injured liver. Within this latent complex, the active TGF-β dimer is non-covalently associated with its propeptide called latency associated peptide (LAP). Only the uncomplexed free TGF-β dimer that has dissociated from LAP can be recruited to its receptor complex.
Binding of LAP/TGF-β to TSP-1 resulting in activation of the cytokine is mediated (at least in non-hepatic tissue) by two sequences within the type 1 repeats: the sequence WxxW, which is present in several copies in TSP-1 and TSP-2, binds directly to the active TGF-β dimer itself and the KRFK motif, which is only present in TSP-1, and binds to the LSKL sequence of LAP, thereby mediating activation of the small latent complex, possibly by changing the conformation of the LAP dimer.17 This process leads to the release of the active TGF-β dimer from the latent complex. Details of this mechanism—for example, how many monomers of TSP-1 (which is generally provided as a homotrimer) bind simultaneously to latent or active TGF-β and if additional cofactors or prior modification(s) such as proteolytic processing are necessary—are still unknown.
We studied expression of TSP-1 in different liver cell types with emphasis on the regulation and action of TSP-1 in cultured rat HSC. In summary, we found that PDGF-BB was a strong inducer of TSP-1 expression in HSC and the data indicate that interactions between TSP-1 and latent or active TGF-β lead to rapid local activation and increased potency of the active TGF-β dimer itself.
MATERIALS AND METHODS
Recombinant human LAP, rat PDGF-BB, human TGF-β1, rat TNF-α, and rat interleukin 6 (IL-6) were purchased from R&D Systems (Minneapolis, Minnesota, USA). KRFK and LSKL peptides were synthesised by Genosys (Sigma, St Louis, Missouri, USA). TSP-1 purified from human platelets was purchased from Sigma. Antibodies against TSP-1 were as follows: Ab2, monoclonal, purchased from Oncogene (Cambridge, Massachusetts, USA), epitope maps to the N terminal half of the central stalk-like region (chymotryptic fragment of 50 kDa); Ab3, monoclonal (Oncogene), epitope maps to the collagen type V binding domain (chymotryptic fragment of 70 kDa); and Ab8, polyclonal (NeoMarkers via Dunn Labortechnik GmbH, Asbach, Germany) was raised against purified full length human platelet TSP-1. Mouse antihuman TGF-β1 neutralising monoclonal antibodies were purchased from Chemicon International (Hampshire, UK). Normal IgG was purchased from Santa Cruz (California, USA).
All animals received humane care in compliance with the German Animal Protection Act, which is in accordance with the National Research Council criteria.
Isolation and culture of HSC, Kupffer cells, and sinusoidal endothelial cells (SEC), and human dermal fibroblasts
Primary HSC were isolated from rat livers by the pronase-collagenase perfusion technique followed by single step density gradient centrifugation with Nycodenz, as described previously.16 Cells were seeded on day 0 with a density of 4×104 cells/cm2 and cultured in Dulbecco’s modification of Eagle’s medium (Bio Whittaker Europe, Verviers, Belgium) containing 4 mM l-glutamine (Flow Laboratories, Bonn, Germany), penicillin (100 IU)/streptomycin (100 μg/ml) (Biochrom KG, Berlin, Germany), and 10% fetal calf serum (FCS; Seromed, Berlin, Germany) under humidified atmosphere of 5% CO2/95% air at 37°C. Fully differentiated MFB were generated by trypsinisation and reseeding of HSC after 7–8 days of primary culture followed by 4–6 days of secondary culture. Kupffer cell and SEC were isolated by centrifugal elutriation. To induce liver fibrosis, prolonged ligation of the common bile duct (BDL) was performed as described previously.18
Human dermal fibroblasts were obtained by outgrowth from skin biopsies of healthy donors. Fibroblasts were cultured as described for HSC but the medium was additionally supplemented with 0.28 mM ascorbic acid.
Isolation of hepatocytes
Hepatocytes were isolated from male Sprague-Dawley rats (strain Han:SPRD from the Institute of Versuchstierkunde, RWTH Aachen, Germany; body weight approximately 250 g) by the two step collagenase method of Seglen.19 Hepatocytes were further purified by centrifugal elutriation, resulting in a final contamination by non-parenchymal cells of <3%.18
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated using the “high pure RNA isolation kit” (Roche, Mannheim, Germany), and under standard conditions equal amounts of each sample were reverse transcribed into cDNA by the action of the AMV reverse transcriptase (Roche). The following primers were used in subsequent PCR (36 cycles for TGF-β1, TSP-1, and rS6, and 27 cycles for PDGF-B): TGF-β1: forward 5′-TGG CGT TAC CTT GGT AAC C-3′, reverse 5′-GGT GTT GAG CCC TTT CCA G-3′; TSP-1: forward 5′-GTG TTT GAC ATC TTT GAA CTC-3′, reverse 5′-CCA AAG ACA AAC CTC ACA TTC-3′; rS6: forward 5′’-GAC TGA CAG ATA CCA CTG TGC CT-3′, reverse 5′-TTA TTT TTG ACT GGA CTC AGA T-3′; PDGF-B: forward 5′-CTG GAG TCG AGT CGG AAA G-3′, reverse 5′-CGA GTT TGA GGT GTC TTG GC-3′. Specificity of PCR products was verified by sequencing (see below). Primers used for sequencing the rat TSP-1 KRFK coding region are indicated in fig 4 ▶.
Figure 4.
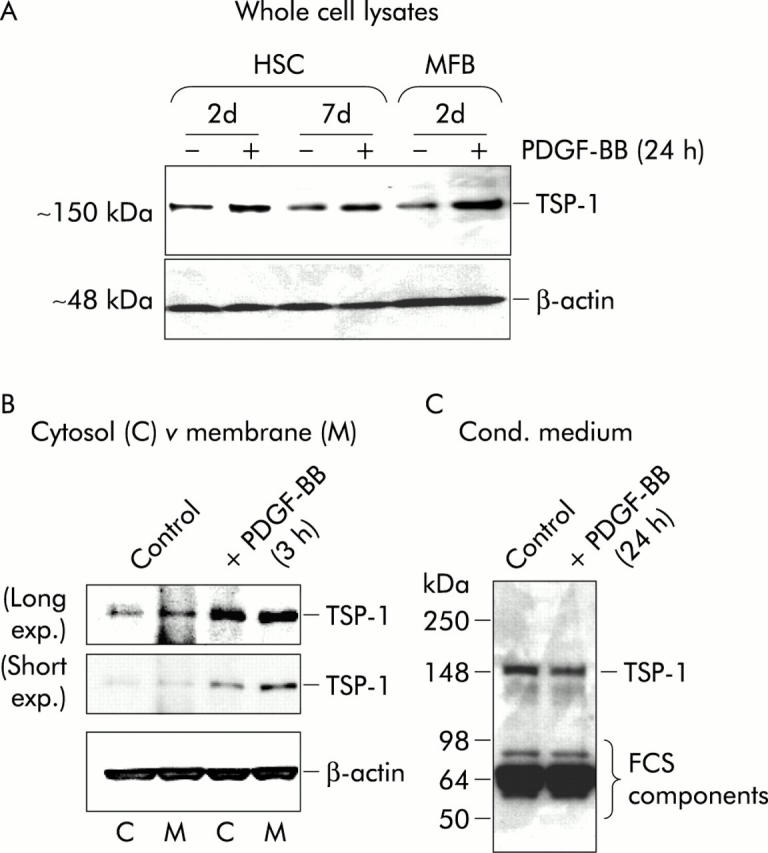
Determination of thrombospondin 1 (TSP-1) protein levels after platelet derived growth factor BB (PDGF-BB) stimulation. Cell lysates and conditioned medium from transdifferentiating cells with (+) or without (−) stimulation with PDGF-BB (20 ng/ml) for 24 hours or three hours, as indicated, were analysed by western blot under reducing conditions using specific mouse monoclonal antibodies against TSP-1. (A) Total cell lysate (15 μg). (B) Cytosol 15 μl (C) or cell membrane (M) sample. (C) Conditioned medium (30 μl) (containing 0.5% fetal calf serum (FCS)) per lane. For (B) and (C), three day old HSC were used. β-Actin expression is shown to demonstrate equal loading. To accentuate differences in band intensities, the blot under (B) is shown with short and long exposure times.
Stimulation with cytokines
Whenever HSC or MFB were stimulated with cytokines, they were serum starved overnight (0.5% FCS), cytokines were added under serum free conditions, and the serum content was supplemented to 0.5% one hour later, remaining at this concentration until the final cell lysis.
Northern hybridisation
High pure RNA was separated using 1% formaldehyde agarose gels (1-5 μg total RNA per lane) and blotted onto nylon membranes (Hybond XL, Amersham Pharmacia Biotech, Buckinghamshire, UK). Hybridisation probes were prepared by labelling 572 bp PCR fragments of rat TSP-1 (primer sequences and RNA isolation as described for RT-PCR above) and human cDNA fragments of TGF-β120 or collagen I α(1)21 with α[32P]dCTP using a random priming kit (Life Technologies, Paisley, Scotland, UK). GAPDH probes were purchased from Sigma Aldrich (Deisenhofen, Germany). Unincorporated nucleotides were removed by ethanol/ammonium acetate precipitation in the presence of excess carrier DNA.
Separation of membrane and cytosolic cell fraction
Cells were pelleted and resuspended in phosphate buffered saline containing 10 μg/ml aprotinin, pepstatin, and leupeptin followed by three cycles of freezing and thawing using a dry ice/ethanol mixture and a 37°C heated water bath. Membrane fragments and cytosolic components were separated by centrifugation (17000 g, 30 minutes, 4°C) and the pellet containing the membrane fragments was dissolved in a volume equal to that of the cytosolic fraction. Sufficient enrichment and separation of the cytosolic and membrane proteins of rat HSC using this method has been demonstrated previously.22
Inhibition of PDGF receptor activity
HSC were isolated and seeded at a density of 4×104 cells/cm2 in the presence or absence of 250 nM of PDGF receptor tyrosine kinase inhibitor I (Calbiochem, Merck Biosciences, Schwalbach/Ts, Germany) dissolved in DMSO. Control cells were cultured only with addition of an equal amount of DMSO. Cells were harvested the next day ( = “day 1”) and TSP-1 and rS6 mRNA levels were investigated by RT-PCR, as described above.
TSP-1 western blot
Cell lysates (20 μg) were separated on 6% sodium dodecyl sulphate polyacrylamide gels under reducing conditions and transferred onto nitrocellulose membranes (Hybond ECL; Amersham) by semi dry transfer system. To detect TSP-1, mouse monoclonal antibodies (Ab-3; 1:300) from Oncogene or mouse monoclonal anti-β-actin antibodies (1:2000) from Sigma were used followed by an enhanced chemiluminescence procedure (with reagents from Amersham).
TGF-β reporter assay
(See also scheme of the experimental set up, as illustrated in fig 6B ▶). Freshly isolated HSC were plated on 96 well plates at a density of 2×104 cells per well. The next day, plating efficiency was determined and the cells were infected with 5×104 viral particles/cell. Adenoviruses were purified using the “Viraprep” kit from Q-Biogene (Heidelberg, Germany) which encode luciferase, driven by multiple copies of a TGF-β response and Smad binding element (“CAGA”), which had originally been isolated from the human PAI promoter.23 On the third day of culture, infected HSC were stimulated for the indicated times, with or without one hour pretreatment with LAP, LSKL, and/or KRFK or antibodies against TSP-1 (25 μg/ml) or TGF-β neutralising antibodies (20 μg/ml). Cell lysis occurred directly in the wells and total luciferase activity per well was measured (using the Steady-Glo Luciferase Assay System; Promega, Madison, Wisconsin, USA) on a liquid scintillation and luminescence counter (1450 Microbeta; Wallac Jet, Turku, Finland). When fibroblasts were used, 5×103 cells per well were seeded followed by the same experimental set up as described for HSC. To compare results of different experiments, all values were normalised to the corresponding (untreated) control.
Figure 6.

CAGA9-MLP-Luc reporter gene studies. (A) Platelet derived growth factor BB (PDGF-BB) treatment increased transforming growth factor β (TGF-β) signalling. One day old hepatic stellate cells (HSC) were infected with adenoviruses encoding a reporter gene (luciferase) whose expression is controlled by multiple copies of a TGF-β response motif (“CAGA9-MLP-Luc”). Two days later, cells were stimulated with PDGF-BB (20 ng/ml) for the indicated time periods, followed by cell lysis and measurement of luciferase activity. Values are mean (SD) (normalised to the control) of two independent experiments, each performed at least in triplicate, per time point. CPS, counts per second. (B) PDGF-BB and TGF-β effects. As illustrated in the scheme above the graph, HSC were infected with adenoviruses and two days after infection, cells were stimulated with the indicated reagents for three hours. TGF-β was used at 0.1 ng/ml as a positive control. Where recombinant latency associated peptide (LAP) (100 ng/ml), LSKL or KRFK peptides (2000 nM), or antibodies against thrombospondin 1 (TSP-1) (Ab2, Ab3, Ab8; 25 μg/ml each) were added together with PDGF-BB (20 ng/ml) or TGF-β (0.1 ng/ml), cells were preincubated with these substances for one hour. As an internal control for experiments where anti-TSP-1 antibodies were used, cells were also incubated with a mixture of normal IgG antibodies (mouse plus rabbit IgG, 10 μg/ml each). Values are mean (SD) (normalised to the corresponding control) of 3–6 ( = n, as indicated within each bar) independent experiments, each performed at least in triplicate per condition ( = at least nine values per condition). *p<0.05 compared with PDGF-BB (P) alone (at 20 ng/ml) or with untreated control (C). NS (P), not significantly different from PDGF-BB alone (at 20 ng/ml). (C) Effects of tumour necrosis factor α (TNF-α) or purified TSP-1. CAGA-luciferase activities after stimulation (three hours) with increasing amounts of TNF-α or purified human TSP-1 were measured and are presented as described in (B).
Sequence analyses
For automated sequencing, the BigDye Terminator v1.1 Cycle Sequencing Kit from Applied Biosystems (Foster City, California, USA) was used on an ABI PRISM 310 Genetic Analyzer according to the manufacturer’s instructions.
If not specified in the legend, all results presented in this study are representative examples of at least two independent experiments ( = two cell preparations from two different animals).
RESULTS
Expression of TSP-1 in different liver cell types
RT-PCR data showed that TSP-1, like TGF-β1, was basally expressed in HSC, MFB, Kupffer cells, and SEC, but not in hepatocytes (fig 1A ▶). Absence of TSP-1 mRNA in hepatocytes was confirmed by northern blot analyses, investigating hepatocyte mRNA from cells isolated from either healthy rat liver from an untreated control animal, from healthy liver 10 days after sham operation, or from injured liver with the beginning of fibrosis, 11 days after BDL (data not shown). TSP-1 mRNA was most highly expressed in freshly cultured HSC (around days 1–3) and remained constant at relatively high levels during further transdifferentiation (fig 1B ▶).
Figure 1.
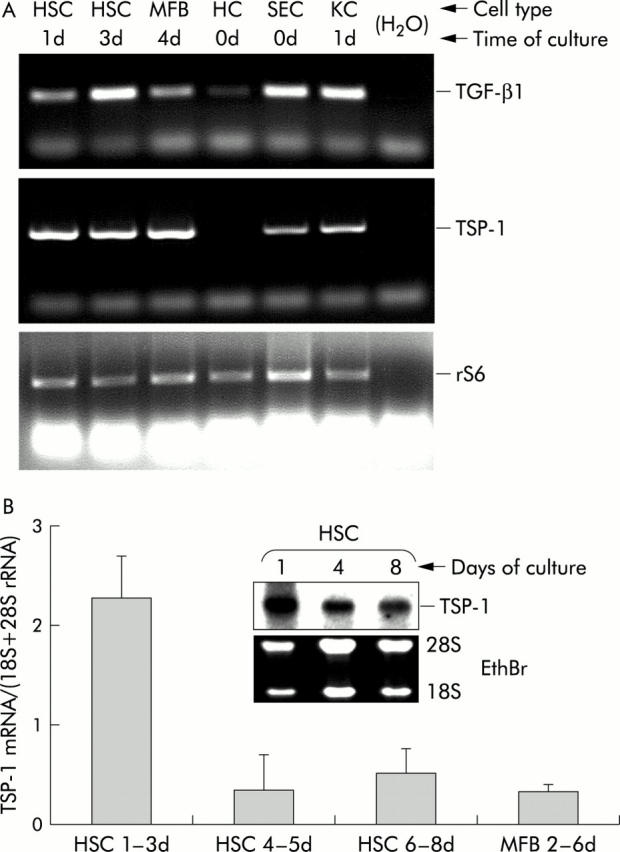
Expression of thrombospondin 1 (TSP-1) mRNA in liver cells. (A) Equal amounts of total RNA from hepatic stellate cells (HSC), myofibroblasts (MFB), hepatocytes (HC), sinusoidal endothelial cells (SEC), and Kupffer cells (KC) either freshly isolated from rat livers (0d) or after the indicated periods of culture, were reverse transcribed into cDNA and expression of transforming growth factor β1 (TGF-β1) and TSP-1 was investigated qualitatively by polymerase chain reaction using specific primers, as described in materials and methods. As internal control, amplification of rS6 is shown. (B) TSP-1 mRNA expression during HSC transdifferentiation. Total RNA was isolated from HSC (primary cultures) or MFB (secondary cultures of HSC) at the indicated time points in culture, and TSP-1 mRNA expression was analysed by northern blotting. The graph shows the results of densitometric analyses of five independent cell preparations, and TSP-1 mRNA values were normalised to 18S and 28S rRNA (mean (SD) from 3–4 individual values per time group). The insert shows one of these blots as a representative example.
PDGF-BB and TNF-α induce coexpression of TSP-1 and TGF-β1 mRNA
To identify possible mediators of TSP expression in activated HSC, cells were stimulated with four different cytokines, each being of known importance during pathological processes such as fibrosis or inflammation. After 24 hours of incubation with either recombinant PDGF-BB, TGF-β1, TNF-α, or IL-6, total RNA was extracted from transdifferentiating HSC and subjected to northern blot analysis. PDGF-BB or TNF-α treatment caused strong upregulation of TSP-1 mRNA (figs 2A ▶, 2B ▶, 3A ▶, 3B ▶) while TSP-2 expression was not affected by these cytokines (data not shown). IL-6 or TGF-β had little or no effect (fig 2A ▶). TSP-1 induction by PDGF-BB was present in quiescent and fully transdifferentiated cells (fig 2A ▶, B) and was dose dependent (fig 2B ▶). To predict possible autocrine PDGF-BB effects during in vitro transdifferentiation, basal expression of PDGF-B mRNA in HSC was investigated by RT-PCR analysis. Interestingly, PDGF-B (like TSP-1) was found to be strongly expressed at early stages of transdifferentiation while much less expression of PDGF-B was detected in fully differentiated cells (fig 2C ▶). These results were confirmed by detailed quantitative analyses of PDGF-B mRNA transcripts (manuscript submitted).
Figure 2.

Analysis of thrombospondin 1 (TSP-1) expression in hepatic stellate cells (HSC) and myofibroblasts (MFB) after cytokine stimulation. (A) Total RNA was prepared after 24 hours of stimulation with platelet derived growth factor BB (PDGF-BB 20 ng/ml), transforming growth factor β1 (TGF-β1 5 ng/ml), interleukin 6 (IL-6 5 ng/ml), or tumour necrosis factor α (TNF-α 10 ng/ml), and by northern blot analysis expression of TSP-1 and TGF-β1 in activated HSC or MFB respectively, was compared with untreated cells. (B) Dose dependency of PDGF-BB or TGF-β1. Total RNA from MFB (3d) was isolated and expression patterns of TSP-1 and TGF-β1 mRNA were investigated by northern blot analysis. Cells were treated with the indicated concentrations of cytokines for four hours. (C) Decreased expression of PDGF-B mRNA during HSC transdifferentiation. Total RNA (1 μg) from one day (1d) or eight day old (8d) HSC and from two day old (2d) MFB was reverse transcribed into cDNA and endogenous expression of PDGF-B mRNA was investigated qualitatively by polymerase chain reaction using specific primers, as described in materials and methods. (D) High TSP-1 expression at early stages of transdifferentiation (day 1) depend on PDGF receptor tyrosin kinase activity. HSC were isolated from healthy rat livers and cultured for one day with or without supplementation of the medium with 250 mM PDGF receptor (PDGFR) tyrosin kinase inhibitor I. Control cultures received only equal amounts of DMSO. Total RNA was isolated and TSP-1 and rS6 mRNA expressions were investigated, as described for fig 1A ▶.
Figure 3.

Analysis of thrombospondin 1 (TSP-1) expression in hepatic stellate cells (HSC) after cytokine stimulation. (A) Time course analysis of transforming growth factor β1 (TGF-β1) and TSP-1 mRNA expressions after stimulation with platelet derived growth factor BB (PDGF-BB), TGF-β1, or tumour necrosis factor α (TNF-α). Total RNA from HSC (2d or 3d) was isolated after the indicated periods of stimulation and expression patterns of TSP-1 and TGF-β1 mRNA were investigated by northern blot analysis. (B) PDGF-BB induced TSP-1 and TGF-β1 mRNA expressions more potently than TNF-α. HSC (3d) were stimulated with either PDGF-BB or TNF-α for three hours and total RNA was isolated and analysed by northern blot analysis. Intensities of the TSP-1 and TGF-β1 signals were quantified densitometrically and normalised to the corresponding 18 and 28 S rRNA. The number of experiments per condition (n) is given within each bar. The graph represents mean values (SD) as fold induction compared with the corresponding untreated controls. Concentration of stimulants as in fig 2A ▶.
To investigate if these high endogenous PDGF-BB levels at early time points might relate to high TSP-1 expression, we plated the cells in the presence of a highly selective cell permeable inhibitor of PDGF receptor tyrosin kinase and indeed TSP-1 mRNA levels on day 1 were clearly reduced under this condition (fig 2D ▶). Moreover, TGF-β1 mRNA expression was coinduced with TSP-1 by the same cytokines, PDGF-BB and TNF-α (figs 2B ▶, 3A ▶, 3B ▶), showing the strongest stimulation as a short time response (∼3 hours of treatment) (figs 2B ▶, 3A ▶). Densitometric analysis of up to eight northern blots per condition showed that PDGF-BB was a more potent inducer of TSP-1 and TGF-β1 mRNA than TNF-α (fig 3B ▶). Stimulation with PDGF-BB for three hours caused an average induction of TSP-1 and TGF-β1 mRNA of ∼8 and ∼5-fold, respectively, while the effect of TNF-α treatment for three hours was only ∼4- and ∼2-fold, respectively.
By western blot analysis, we showed that induction of TSP-1 mRNA by PDGF-BB correlated with increasing amounts of TSP-1 protein in cell lysates of transdifferentiating cells stimulated for 24 hours (fig 4A ▶). The degree of stimulation varied between different time points during the transdifferentiation process with a tendency to be more pronounced at later stages. To further analyse the cellular localisation of TSP-1 after PDGF stimulation, we prepared cytosolic and membrane protein fractions according to a method that has recently been shown by us to separate such proteins effectively using primary HSC.22 As shown in fig 4B ▶, TSP-1 protein in the cytosol and especially in the membrane increased after only three hours of stimulation. In contrast, in conditioned medium of HSC cultures, no changes in TSP-1 were detectable by western blot after 24 hours of stimulation (fig 4C ▶) indicating that there was no increase in soluble TSP-1.
Sequence analysis of rat TSP-1
TSP-1 is proposed as an activator of latent TGF-β due to interaction of TSP-1 with a defined amino acid sequence of LAP.17 Therefore, a 191 bp region of rat TSP-1 cDNA was sequenced and compared with the corresponding human and mouse sequences (fig 5 ▶). We demonstrated that the domain encoding for the KRFK motif, which is believed to be necessary for activation of latent TGF-β (see introduction for details), is highly conserved among all three species, confirming the potency of rat TSP-1 to regulate the activity of TGF-β at the molecular level.
Figure 5.

Sequence alignment of thrombospondin 1 (TSP-1) from rat, human, and mouse: 191 bp of rat TSP-1 cDNA was sequenced using the indicated primers and the resulting fragment was compared with known sequences of human (gi4507484) and mouse (gi202196) TSP-1. The region encoding the transforming growth factor β activating motif KRFK is identical in all three species (box). Single base pairs that differ in the rat and/or human sequence compared with the mouse sequence are underlined.
Measurement of TGF-β signalling
To further analyse the effect of PDGF-BB or TNF-α induced TSP-1 expression on activation of latent TGF-β and subsequent TGF-β/Smad signalling, HSC were infected with recombinant adenoviruses encoding a reporter gene (luciferase) which is driven by multiple copies of a TGF-β response motif (“CAGA”, see materials and methods for details). Luciferase activities of control cultures or after increasing periods of stimulation with PDGF-BB were measured in cell lysates and a gradual increase in luciferase activity indicated that after PDGF-BB treatment, active TGF-β was generated from endogenous (latent) sources (fig 6A ▶). The effect became detectable after only 1–3 hours of stimulation, reached a maximum at 4–5 hours, and declined thereafter (fig 6A ▶). The nature of the PDGF effect was analysed in more detail after three hours of stimulation either with or without one hour pretreatment of the cells with certain inhibitors (see scheme on top of fig 6B ▶ for details of the experimental set up). As a positive internal control, cells were treated with a small concentration (0.1 ng/ml) of recombinant active TGF-β1 for three hours which led to an average increase in luciferase activity of approximately threefold, demonstrating the sensitivity of this assay (fig 6B ▶). PDGF-BB dependent luciferase activity was inhibited in the presence of excessive amounts of recombinant LAP or by addition of a short synthetic peptide (LSKL), specifically covering the binding site of TSP-1 (KRFK) for LAP or by antibodies against TSP-1 (fig 6B ▶). Two of the TSP-1 antibodies used here (Ab2 and Ab3) are monoclonals that bind to a region of TSP-1 which includes the KRFK motif and both reduce the PDGF-BB effect, with Ab3 being more potent than Ab2. The third antibody against TSP-1, which was used in this context (Ab8), is a polyclonal which was generated by immunisation with the purified full length protein and this antibody completely inhibits PDGF induced TGF-β signalling. Using only the TSP-1 fragment, KRFK, stimulation of luciferase activity was also achieved but to a lesser extent compared with PDGF-BB (fig 6B ▶).
Human TSP-1 purified from platelets was only effective at the highest concentration tested, 100 ng/ml (fig 6C ▶). The stronger effect of PDGF-BB may result from a parallel increase in the synthesis of endogenous TGF-β. TNF-α, in spite of its TSP-1 inducing ability, failed to stimulate CAGA-luciferase activity (fig 6C ▶). When active TGF-β was combined with PDGF-BB, superinduction of luciferase activity occurred, which again was blocked by anti-TSP-1 antibodies (Ab8) or LSKL (fig 7A ▶). Pretreatment of cells with TGF-β neutralising antibodies blocked the effects of TGF-β or PDGF-BB alone and also completely inhibited luciferase superinduction after stimulation with both cytokines together (fig 7A ▶). Furthermore, Ab8 or LSKL decreased signal transduction of recombinant active TGF-β itself (fig 7A ▶). To our knowledge, this is the first report describing such a direct effect of TSP-1 on active TGF-β, and therefore we further investigated if this was a cell type specific effect or a more general mechanism. Using human dermal fibroblasts instead of HSC, similar results were achieved and we demonstrated that fibroblasts basally expressed significant levels of TSP-1 (fig 7B ▶, C). These results indicate that endogenous TSP-1 is not only involved in the activation of latent TGF-β but also enhances the effectiveness of the active TGF-β dimer itself. However, a combination of active TGF-β with reasonable amounts of human TSP-1 purified from platelets did not superinduce the TGF-β effect (fig 7A ▶). As expected, a mixture of KRFK and LSKL did not show any effect (figs 6B ▶, 7A ▶).
Figure 7.

CAGA9-MLP-Luc reporter gene studies. (A) Effects of transforming growth factor β (TGF-β) alone or in combination with platelet derived growth factor BB (PDGF-BB). CAGA-luciferase activities after stimulation (three hours) with either TGF-β alone (0.1 ng/ml) or in combination with PDGF-BB (5 ng/ml) were measured in hepatic stellate cells, and are presented as described in fig 6B ▶. The number of experiments performed per condition (n) is given below the columns. Peptides (2000 nM) or antibodies (25 μg/ml) were added one hour prior to the indicated cytokine(s) and the values in parentheses indicate the amounts used (in ng/ml). Pretreatment of cells with TGF-β neutralising antibodies (20 μg/ml) strongly inhibited reporter gene activation. (B) CAGA-luciferase activities after stimulation (three hours) with TGF-β (0.1 or 0.5 ng/ml) were measured in human dermal fibroblasts, and are presented as described in fig 6B ▶. (C) Western blot of cell lysates (10 μg per lane) from human dermal fibroblasts of three different healthy donors, indicating that these cells express relatively high basal levels of thrombospondin 1 (TSP-1) compared with rat myofibroblasts (MFB). β-actin expression is shown as a loading control. For the CAGA-luciferase assays shown in (B), fibroblasts from donor No 1 were used and the graph shows the average of four independent experiments, each performed in triplicates (SD).
Recombinant LAP alone led to a moderate decrease in the control (basal) value of luciferase activity (fig 6B ▶), indicating the presence of small basal amounts of active TGF-β in cultured HSC (autocrine stimulation).
DISCUSSION
We have demonstrated that TSP-1 is expressed during culture activation of HSC with the highest levels at the early time points. Freshly isolated parenchymal cells (hepatocytes) from healthy rats did not express TSP-1. A similar expression pattern was found for TGF-β1 and its binding protein LTBP.16 Kupffer cells, SEC, and quiescent HSC were identified as cellular sources of TSP-1 and all three cell types were confirmed as TGF-β1 producers.24,25
TGF-β is secreted almost exclusively as a latent molecule which needs to be converted into its active form to become functional.26 According to the findings in other tissues/cell types11,27 and recently in rat liver,15 TSP-1 is thought to activate latent TGF-β1 in HSC, thereby promoting TGF-β mediated fibrogenesis. Only a few details of the activation mechanism are currently known. The amino acid motif “KRFK”, which is responsible for the binding of TSP-1 to the latent TGF-β complex and for its activation,15,17 was identified in the rat TSP-1 protein sequence.
In vivo, PDGF-BB is released from activated HSC or by infiltrating inflammatory cells during liver injury. Cross talk between intracellular pathways leading to proliferation and migration of cultured HSC is stimulated by PDGF-BB.28,29 In HSC, the highest PDGF-B mRNA levels were found at the early stages of transdifferentiation, which was strongly correlated with the expression profile of TSP-1, and blockage of PDGF receptor tyrosin kinase activity during initiation of transdifferentiation using a highly specific cell permeable inhibitor clearly reduced TSP-1 expression. Hence PDGF-BB and, to a minor extent, TNF-α, were found to be strong inducers of TSP-1 expression in activated HSC while both did not affect TSP-2 expression (data not shown). Transient induction of TSP-1 by PDGF-BB was previously reported using growth arrested rat smooth muscle cells, suggesting a role for TSP-1 in the proliferative response of these cells to PDGF.30 In the presence of actinomycin D, PDGF-mediated increases in TSP-1 can be blocked,30,31 implying that PDGF regulates TSP message at the level of transcription. Furthermore, proliferating mesangial cells express TSP-1 de novo in experimentally induced nephritis and it was shown that treatment of animals with anti-PDGF and anti-bFGF antibodies resulted in downregulation of TSP-1 expression, indicating regulation by PDGF and bFGF.32 Additionally, PDGF and TSP-1 were identified as autocrine motility factors that enhance smooth muscle cell migration, and it was found that the effect of PDGF is indirect, by inducing TSP-1 expression,33 indicating that PDGF induced TSP-1 expression is involved in the formation of artherosclerotic lesions at sites of vascular injury and/or inflammation. Apart from PDGF, other growth factors have been found to induce TSP-1 expression and may be functional in liver fibrogenesis dependent TSP-1 expression. For example, angiotensin II increased TSP-1 expression in human mesangial cells within six hours by activating p38 MAPK and JNK signalling.34
Parallel with coinduction of TSP-1 and TGF-β1 mRNA expression (within 1–5 hours), PDGF-BB treatment led to increased (CAGA)9-MLP-Luc reporter gene activation in HSC. As (CAGA)9-MLP-Luc is solely induced by binding of activated Smad3/4 complexes, the effect must be mediated by PDGF-BB stimulated TGF-β signalling. PDGF-BB dependent induction of TGF-β signalling was inhibited by pretreatment of the cells with a TGF-β neutralising antibody or by obstructing the binding site of TSP-1 for TGF-β/LAP with LSKL peptides or by preincubation with anti-TSP-1 antibodies, confirming that this effect is TGF-β dependent and mediated by TSP-1.
In contrast with PDGF-BB, TNF-α did not significantly induce (CAGA)9-MLP-Luc activation, which may be explained by a very moderate increase in TGF-β (∼2-fold compared with ∼5-fold by PDGF-BB) and TSP-1 mRNA (∼4-fold compared with ∼8-fold by PDGF-BB). In addition, TNF-α has been reported to be an antagonist of TGF-β/Smad signalling by downregulating TGF-β receptor II,35 by upregulating AP-1 which prevents Smad3-DNA interactions,36 or by upregulating Smad7.37 However, such mechanisms do not seem to contribute to the observed lack of reporter gene induction by TNF-α in HSC because the effects of TGF-β and PDGF-BB are not decreased by costimulation with TNF-α (data not shown). So far, we have no explanation for these results but we speculate that together with TSP-1, induction of cofactors such as TSP-1 receptor(s) at the cell surface or certain proteases might be necessary to induce (CAGA)9-MLP-Luc activity and that these are induced by active TGF-β or PDGF-BB but not by TNF-α.
When added in combination with recombinant active TGF-β, PDGF-BB caused “superinduction” of (CAGA)9-MLP-Luc which was almost completely blocked in the presence of TSP-1 neutralising agents (Ab8 or LSKL). This indicates that PDGF-BB dependent TSP-1 expression, apart from activation of latent TGF-β, has a direct promoting effect on active TGF-β. This is supported by the finding that neutralisation of endogenous TSP-1 in HSC is sufficient to inhibit signalling of exogenously added active TGF-β. A similar influence of TSP-1 on active TGF-β was identified in human dermal fibroblasts, indicating a more general mechanism. Interestingly, a physical interaction of TSP-1 with PDGF has been reported, leading to stabilisation of this cytokine, thereby potentiating its effects,38,39 and it might be speculated that TSP-1 has similar functions for TGF-β.
Previous reports identified the KRFK peptide of TSP-1 as sufficient to activate latent TGF-β.17 In line with this, addition of this peptide to HSC cultures increased reporter gene activity whereas the LAP specific LSKL peptide was able to abrogate the effects of TSP-1. In addition, LSKL displayed a negative regulatory impact on the activated TGF-β dimer itself, in HSC and fibroblasts. In a model of TSP-1 latent TGF-β interactions, three WxxW motifs in the neighbourhood of the KRFK sequence bound mature TGF.12,17 Therefore, we conclude that a direct interaction of TSP-1, possibly via the identified WxxW motifs, has a stimulatory function for active TGF-β and that excessive amounts of LAP/LSKL or anti-TSP-1 antibodies decrease TGF-β effects by physically interfering with this mode of binding.
We attempted to mimic the effects of PDGF-BB by directly adding TSP-1 purified from human platelets to HSC. According to a quantitative investigation of TSP-1 synthesis in malignant glioma cells,40 reasonable amounts synthesised within three hours should be in the range of approximately 20 ng/ml. However, concentrations as high as 100 ng/ml were needed to achieve a twofold increase in luciferase activity in HSC. The different efficiencies of endogenously synthesised TSP-1 and exogenously added human TSP-1 from platelets may be due to post translational modifications such as enzymatic processings of TSP-1 or the association with cofactors, which might not be present in platelets or were lost during purification. Furthermore, we observed that purified TSP-1 is very unstable and degrades within 30 minutes at 37°C (data not shown).
From our observations, we conclude that in fibrotic liver, PDGF-BB stimulates HSC or MFB (and perhaps also Kupffer cells and SEC) to produce large amounts of TSP-1 which in turn enhance profibrogenic TGF-β activity. This is shown by the ability of TSP-1 to mediate rapid local activation of latent TGF-β in HSC and to enhance the efficiency of signalling by active TGF-β. This does not exclude the possibility that additional factors (that is, enzymes such as plasmin41 or tissue transglutaminase)42 or the interaction of TGF-β or TSP-1 with receptors such as integrins43 or the M6P/IGF-II receptor,44 may play a role. Tissue transglutaminase has recently been shown by us to be present in the membranes of HSC and to be upregulated during transdifferentiation.22 However, identification of TSP-1 as a significant profibrogenic TGF-β promoting factor during activation of HSC adds important new information for the development of antifibrotic therapies. The possibility of inhibiting profibrogenic TGF-β signalling via TSP-1 has successfully been used in experimental inflammatory kidney disease.45 In these experiments, antisense oligonucleotides targeting TSP-1 markedly abrogated de novo synthesis of TSP-1 and subsequent decreased activation but not expression of TGF-β. In the liver, injection of the LSKL peptide to rats strongly reduced dimethylnitrosamine induced organ damage and fibrosis.15 Finally, the stimulating effect of TSP-1 on active TGF-β described in the present study represents a new regulatory mechanism that is found in different cell types and may therefore be important in many different tissues apart from the liver.
Acknowledgments
The authors are grateful to U Klußmann, CG Tag, and S Sauer-Lehnen for technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 542: Molekulare Grundlagen Zytokin-gesteuerter Entzündungsprozesse: Signaltransduktion und pathophysiologische Konsequenzen, Aachen) and the Dietmar Hopp Stiftung GmbH.
Abbreviations
TGF-β, transforming growth factor β
PDGF-BB, platelet derived growth factor BB
TSP, thrombospondin
HSC, hepatic stellate cells
MFB, myofibroblasts
SEC, sinusoidal endothelial cell
TNF-α, tumour necrosis factor α
IL-6, interleukin 6
LAP, latency associated peptide
LTBP, latent TGF-β binding protein
RT-PCR, reverse transcription-polymerase chain reaction
FCS, fetal calf serum
BDL, bile duct ligation
Conflict of interest: None declared.
REFERENCES
- 1.Bornstein P . Thrombospondins as matricellular modulators of cell function. J Clin Invest 2001;107:929–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iruela-Arispe ML, Liska DJ, Sage EH, et al. Differential expression of thrombospondin 1, 2 and 3 during murine development. Dev Dyn 1993;197:40–56. [DOI] [PubMed] [Google Scholar]
- 3.Tooney PA, Sakai T, Sakai K, et al. Restricted localization of thrombospondin-2 protein during mouse embryogenesis: a comparison to thrombospondin-1. Matrix Biol 1998;17:131–43. [DOI] [PubMed] [Google Scholar]
- 4.Kyriakides TR, Zhu YH, Smith LT, et al. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol 1998;140:419–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lawler J, Sunday M, Thibert V, et al. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Invest 1998;101:982–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donoviel DB, Framson P, Eldridge CF, et al. Structural analysis and expression of the human thrombospondin gene promoter. J Biol Chem 1988;263:18590–3. [PubMed] [Google Scholar]
- 7.Laherty CD, Gierman TM, Dixit VM. Characterization of the promoter region of the human thrombospondin gene. DNA sequences within the first intron increase transcription. J Biol Chem 1989;264:11222–7. [PubMed] [Google Scholar]
- 8.Adolph KW, Liska DJ, Bornstein P. Analysis of the promoter and transcription start sites of the human thrombospondin 2 gene (THBS2). Gene 1997;193:5–11. [DOI] [PubMed] [Google Scholar]
- 9.Reed MJ, Iruela-Arispe L, O’brien ER, et al. Expression of thrombospondins by endothelial cells. Injury is correlated with TSP-1. Am J Pathol 1995;147:1068–80. [PMC free article] [PubMed] [Google Scholar]
- 10.Kyriakides TR, Tam JW, Bornstein P. Accelerated wound healing in mice with a disruption of the thrombospondin 2 gene. J Invest Dermatol 1999;113:782–7. [DOI] [PubMed] [Google Scholar]
- 11.Crawford SE, Stellmach V, Murphy-Ullrich JE, et al. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 1998;93:1159–70. [DOI] [PubMed] [Google Scholar]
- 12.Murphy-Ullrich JE, Poczatek M. Activation of latent TGF-β by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev 2000;11:59–69. [DOI] [PubMed] [Google Scholar]
- 13.El-Youssef M, Mu Y, Huang L, et al. Increased expression of transforming growth factor-beta1 and thrombospondin-1 in congenital hepatic fibrosis: possible role of the hepatic stellate cell. J Pediatr Gastroenterol Nutr 1999;28:386–92. [DOI] [PubMed] [Google Scholar]
- 14.Hayashi K, Kurohiji T, Shirouzu K. Localization of thrombospondin in hepatocellular carcinoma. Hepatology 1997;25:569–74. [DOI] [PubMed] [Google Scholar]
- 15.Kondou H, Mushiake S, Etani Y, et al. A blocking peptide for transforming growth factor-β1 activation prevents hepatic fibrosis in vivo. J Hepatol 2003;39:742–8. [DOI] [PubMed] [Google Scholar]
- 16.Breitkopf K, Lahme B, Tag CG, et al. Expression and matrix deposition of latent transforming growth factor β binding proteins in normal and fibrotic rat liver and transdifferentiating hepatic stellate cells in culture. Hepatology 2001;33:387–96. [DOI] [PubMed] [Google Scholar]
- 17.Schultz-Cherry S, Chen H, Mosher DF, et al. Regulation of TGF-β activation by discrete sequences of thrombospondin 1. J Biol Chem 1995;270:7304–10. [DOI] [PubMed] [Google Scholar]
- 18.Kountouras J, Billing BH, Scheuer PJ. Prolonged bile duct obstruction: a new experimental model for cirrhosis in the rat. Br J Exp Pathol 1984;65:305–11. [PMC free article] [PubMed] [Google Scholar]
- 19.Seglen PO. Isolation of hepatocytes by collagenase perfusion. In: Tyson CA, Frazier JM, eds. Methods in toxicology. New York: Academic Press, 1993:231–43.
- 20.Qian SW, Kondaiah P, Roberts AB, et al. cDNA cloning by PCR of rat transforming growth factor β-1. Nucleic Acids Res 1990;18:3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chu ML, Myers JC, Bernard MP, et al. Cloning and characterization of five overlapping cDNAs specific for the human pro α1(I) collagen chain. Nucleic Acids Res 1982;10:5925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schnabel C, Sawitza I, Tag CG, et al. Expression of cytosolic and membrane associated tissue transglutaminase in hepatic stellate cells and its upregulation during transdifferentiation to myofibroblasts in culture. Hepatol Res 2004;28:140–5. [DOI] [PubMed] [Google Scholar]
- 23.Dennler S, Itoh S, Vivien D, et al. Direct binding of Smad3 and Smad4 to critical TGF β-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J 1998;17:3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roth S, Gong W, Gressner AM. Expression of different isoforms of TGF-β and the latent TGF-β binding protein (LTBP) by rat Kupffer cells. J Hepatol 1998;29:915–22. [DOI] [PubMed] [Google Scholar]
- 25.Bissell DM, Wang SS, Jarnagin WR, et al. Cell-specific expression of transforming growth factor-β in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 1995;96:447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koli K, Saharinen J, Hyytiainen M, et al. Latency, activation, and binding proteins of TGF-β. Microsc Res Tech 2001;52:354–62. [DOI] [PubMed] [Google Scholar]
- 27.Hugo C, Kang DH, Johnson RJ. Sustained expression of thrombospondin-1 is associated with the development of glomerular and tubulointerstitial fibrosis in the remnant kidney model. Nephron 2002;90:460–70. [DOI] [PubMed] [Google Scholar]
- 28.Yang C, Zeisberg M, Mosterman B, et al. Liver fibrosis: insights into migration of hepatic stellate cells in response to extracellular matrix and growth factors. Gastroenterology 2003;124:147–59. [DOI] [PubMed] [Google Scholar]
- 29.Pinzani M . PDGF and signal transduction in hepatic stellate cells. Front Biosci 2002;7:d1720–6. [DOI] [PubMed] [Google Scholar]
- 30.Majack RA, Cook SC, Bornstein P. Platelet-derived growth factor and heparin-like glycosaminoglycans regulate thrombospondin synthesis and deposition in the matrix by smooth muscle cells. J Cell Biol 1985;101:1059–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Majack RA, Mildbrandt J, Dixit VM. Induction of thrombospondin messenger RNA levels occurs as an immediate primary response to platelet-derived growth factor. J Biol Chem 1987;262:8821–5. [PubMed] [Google Scholar]
- 32.Hugo C, Pichler R, Meek R, et al. Thrombospondin 1 is expressed by proliferating mesangial cells and is up-regulated by PDGF and bFGF in vivo. Kidney Int 1995;48:1846–56. [DOI] [PubMed] [Google Scholar]
- 33.Yabkowitz R, Mansfield PJ, Ryan US, et al. Thrombospondin mediates migration and potentiates platelet-derived growth factor-dependent migration of calf pulmonary artery smooth muscle cells. J Cell Physiol 1993;157:24–32. [DOI] [PubMed] [Google Scholar]
- 34.Naito T, Masaki T, Nikolic-Paterson DJ, et al. Angiotensin II induces thrombospondin-1 production in human mesangial cells via p38 MAPK and JNK: a mechanism for activation of latent TGF-beta1. Am J Physiol Renal Physiol 2004;286:278–87. [DOI] [PubMed] [Google Scholar]
- 35.Yamane K, Ihn H, Asano Y, et al. Antagonistic effects of TNF-α on TGF-β signaling through down-regulation of TGF-β receptor type II in human dermal fibroblasts. J Immunol 2003;171:3855–62. [DOI] [PubMed] [Google Scholar]
- 36.Verrecchia F, Pessah M, Atfi A, et al. Tumor necrosis factor-α inhibits transforming growth factor-beta/Smad signaling in human dermal fibroblasts via AP-1 activation. J Biol Chem 2000;275:30226–31. [DOI] [PubMed] [Google Scholar]
- 37.Bitzer M, von Gersdorff G, Liang D, et al. A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev 2000;14:187–97. [PMC free article] [PubMed] [Google Scholar]
- 38.Hogg PJ, Hotchkiss KA, Jimenez BM, et al. Interaction of platelet-derived growth factor with thrombospondin 1. Biochem J 1997;326:709–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krishnaswami S, Ly QP, Rothman VL, et al. Thrombospondin-1 promotes proliferative healing through stabilization of PDGF. J Surg Res 2002;107:124–30. [DOI] [PubMed] [Google Scholar]
- 40.Amagasaki K, Sasaki A, Kato G, et al. Antisense-mediated reduction in thrombospondin-1 expression reduces cell motility in malignant glioma cells. Int J Cancer 2001;94:508–12. [DOI] [PubMed] [Google Scholar]
- 41.Okuno M, Akita K, Moriwaki H, et al. Prevention of rat hepatic fibrosis by the protease inhibitor, camostat mesilate, via reduced generation of active TGF-β. Gastroenterology 2001;120:1784–800. [DOI] [PubMed] [Google Scholar]
- 42.Kojima S, Nara K, Rifkin DB. Requirement for transglutaminase in the activation of latent transforming growth factor-beta in bovine endothelial cells. J Cell Biol 1993;121:439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Munger JS, Huang X, Kawakatsu H, et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999;96:319–28. [DOI] [PubMed] [Google Scholar]
- 44.de Bleser PJ, Jannes P, van Buul-Offers SC, et al. Insulin like growth factor-II/mannose 6-phosphate receptor is expressed on CCl4-exposed rat fat-storing cells and facilitates activation of latent transforming growth factor-beta in cocultures with sinusoidal endothelial cells. Hepatology 1995;21:1429–37. [DOI] [PubMed] [Google Scholar]
- 45.Daniel C, Takabatake Y, Mizui M, et al. Antisense oligonucleotides against thrombo-spondin-1 inhibit activation of TGF-β in fibrotic renal disease in the rat in vivo. Am J Pathol 2003;163:1185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]