

Abstract
Background and aims: O6-methylguanine methyltransferase (MGMT) repairs inappropriately methylated guanine in DNA. MGMT mutations have not previously been reported in cancers, but in colorectal tumours MGMT promoter methylation is common and has been associated with increased G:C>A:T transitions, a high frequency of K-ras mutations, and low level microsatellite instability (MSI low). However, some have suggested that MGMT changes are background or secondary events, with little importance for tumorigenesis.
Methods: We have analysed fresh frozen colorectal cancers and colorectal cancer cell lines for MGMT changes: mutations, allelic loss, and protein expression.
Results: Six of 113 cancers harboured somatic missense MGMT mutations, at least three of which probably caused reduced MGMT function and were accompanied by silencing or loss of the wild-type allele. Cancers with pathogenic MGMT mutations tended to harbour G:C>A:T somatic mutations at other loci. Overall, MGMT expression was reduced or lost in more than half of the cancers. We found no association between MGMT expression and the somatic mutation spectrum at APC, beta-catenin, K-ras, or p53, but decreased MGMT expression was weakly associated with the presence of a G:C>A:T change at any one of these loci. Reduced MGMT expression was not however associated with an increased frequency of K-ras mutations or with MSI low.
Conclusion: In summary, we found that mutation of MGMT contributes to decreased protein function. Our findings provide good evidence to show that MGMT changes, including methylation, are selected rather than background events, at least in some cases. Decreased MGMT expression or function probably has a weak or moderate effect on the mutation spectrum in colorectal cancers.
Keywords: O6-methylguanine methyltransferase, colorectal cancer, G>A transitions, MGMT, methylation, mutation
O6-methylguanine DNA methyltransferase (MGMT) acts to repair inappropriately methylated guanine residues in DNA. In the absence of MGMT activity, O6-methylguanine (MeG) mispairs with thymine during DNA replication, potentially resulting in G:C>A:T transitions.1 MGMT catalyses the transfer of the methyl group from the DNA to an active site cysteine residue of the MGMT protein, resulting in restoration of the DNA structure in a single step. The S-methylcysteine formed at the active site of MGMT is not converted back to cysteine and the protein can therefore only act once. The methylated form of the MGMT protein undergoes a conformational change which leads to its rapid degradation.2 MGMT can also act to remove adducts larger than methyl groups from guanine. In vitro experiments using mammalian cell lines have shown that endogenous MGMT expression protects them from spontaneous G:C>A:T transitions in the APRT gene.1 Transgenic mice overexpressing Mgmt are protected against G:C>A:T mutations in aberrant colorectal crypt foci.3 It has been suggested, furthermore, that loss of MGMT expression confers sensitivity to alkylating agents in cancer therapy.4
Hypermethylation of the MGMT promoter has been recognised for several years as a cause of MGMT transcriptional silencing in cell lines and cancers defective in MeG repair.5,6,7,8,9,10,11,12,13 It has been postulated that lack of MGMT expression increases the spontaneous G:C>A:T mutation rate in tumours in vivo.1,14 This may result either from underlying differences between MGMT activity in normal bowel15 or from somatic changes acquired during tumorigenesis.
MGMT inactivation has specifically been implicated in colorectal tumorigenesis.16–18 Research has focused on silencing of MGMT transcription by promoter methylation as the cause of decreased protein expression which has been detected using western analysis and/or immunohistochemistry. MGMT methylation appears to be associated with loss of nuclear protein expression in tissues, although the concordance is typically imperfect; for example, concordance of about 75% was reported by Whitehall and colleagues.19 The consequences of MGMT deficiency have been tested by studying mutation spectra in the K-ras and p53 genes. Esteller and colleagues20 reported that MGMT methylation was associated with an increased frequency of K-ras mutations, in particular G:C>A:T transitions; MGMT protein expression was not, however, determined by this study. Esteller and colleagues18 subsequently also showed an association in colorectal tumours between MGMT methylation and G:C>A:T mutations in p53. Such associations have not however been universally reported, with Laiho and colleagues21 showing no link between MGMT methylation and K-ras mutations in a set of colorectal cancers.
Whitehall et al19 found MGMT methylation to be associated not only with a high frequency of K-ras mutation and a tendency to G:C>T:A transitions but also with low level microsatellite instability (MSI low) in colorectal tumours. It was not clear from this study whether or not MGMT expression was similarly associated with MSI low. It was hypothesised that MSI low arose as a result of overload of the DNA mismatch repair system by G:C>T:A mutations. Laiho et al however found no evidence for a relationship between MGMT promoter methylation and the frequency of microsatellite mutations.21
A review of the evidence concerning MGMT in colorectal tumours suggests that its role remains uncertain in some respects. Firstly, it is striking that most associations of MGMT changes with somatic mutations have focused on promoter methylation rather than protein expression, even though the latter would be expected to be a better indication of protein function; promoter methylation might, for example, occur as part of the CpG island methylator phenotype (CIMP) rather than as a result of selection for functional changes.22 Secondly, most studies of MGMT methylation have used methylation specific polymerase chain reaction (PCR), a sensitive but potentially non-specific technique which may detect partial rather than complete loss of gene expression. Thirdly, very little work has been done looking for somatic mutations or allelic loss at MGMT in sporadic cancers, despite the fact that the topology and overall structure of MGMT is highly conserved, particularly in the C terminal domains. Identification of mutations would greatly strengthen the case that MGMT changes are selected, rather than background or secondary events in tumorigenesis.23 Fourthly, the relationship between MGMT expression and MSI low remains unproven. In this study, we screened a set of fresh frozen unselected sporadic cancers of the colorectum and colorectal cancer cell lines for mutations in and allelic loss at MGMT, and determined protein expression using immunohistochemistry or western analysis as appropriate. We have compared our findings with other mutations (APC, beta-catenin, K-ras, and p53) present in each cancer and with phenotypic features such as MSI and chromosomal instability (CIN).
METHODS
Frozen samples of 81 colorectal carcinomas and paired normal colon from a sequential series were taken from colectomy specimens from St Mark’s Hospital, London. Histological review showed that all haematoxylin and eosin stained sections taken contemporaneously from archival sections from the same tumour consisted of >75% carcinoma. Cases with inflammatory bowel disease or a known Mendelian cancer syndrome were excluded. For each tumour, the following clinicopathological data were collected: patient age, Dukes’ stage, grade, and anatomical site in the colon. A panel of 32 colorectal cell lines (C10, C125, COLO320, HCT8, VACO4S, C32, COLO678, LS174T, VACO5, C70, SW948, LS180, PC/JW, SW1116, SW480, DLD1/HCT15, HT29, HRA19, C80, SW620, GP2D, SW1222, SKCO1, C84, COLO201, GP5D, HT55, VACO10, LIM1863, SW48, LOVO, SW403) was also studied. No fresh frozen cancer or cell line had been subjected to alkylating agent therapy. DNA was extracted from all samples and mRNA and protein were extracted from the cell lines, all using standard methods.
Fluorescent-single strand conformational polymorphism (F-SSCP) analysis was performed to screen for variants in the coding regions and exon-intron boundaries of the MGMT gene. Oligonucleotides and reaction conditions used to amplify each fragment are available from the authors. Samples were run at 18°C and 24°C on the ABI3100 capillary sequencer. All tumours with bandshifts on F-SSCP analysis were sequenced in forward and reverse orientations for that exon using a new unlabelled PCR product, the ABI Big Dye Terminator Ready Reaction Mix, and the ABI 377 semi automated sequencer. All sequencing reactions were performed alongside the paired normal DNA sample.
Some of the colorectal cancer cell lines, including those showing mutations in MGMT, were screened for MGMT mRNA expression using reverse transcription-PCR. A segment of MGMT cDNA was amplified using the following oligonucleotides 5′-TGG AGC TGT CTG GTT GTG AG-3′ and 5′-CTG GTG AAC GAC TCT TGC TG-3′. Details of analysis and PCR reaction conditions are available from the authors. The cDNA was then sequenced in forward and reverse directions to determine the presence or absence of the wild-type and mutant alleles.
Loss of heterozygosity (LOH, allelic loss) analysis at MGMT was performed on the 96 colorectal carcinomas at four microsatellite markers (D10S1703, D10S214, D10S1588, and D10S1222) located approximately 1.5–4 Mb from the MGMT locus using standard protocols, dye labelled oligonucleotides, and the ABI377 sequencer. Allelic loss was scored if the dosage of one allele in the tumour decreased by 50% or more relative to the other allele, after correcting for the relative allele peak areas in constitutional DNA. LOH in the colorectal cancer cell lines was inferred, where possible, by direct inspection of sequence electropherograms (for example, in samples with mutations within the MGMT gene).
We screened for mutations in APC, beta-catenin (exon 3), K-ras (exon 1), and p53, as previously reported.24 All samples had previously been scored for MSI25 and CIN.26
MGMT protein expression was assessed in fresh frozen cancers using immunohistochemistry. Serial sections (4 μm) from fixed samples taken at the same time as the frozen samples were deparaffinised, rehydrated, and subjected to antigen retrieval (pressure cooking two minutes at full pressure in 3 litres of 0.01 M citrate buffer). Sections were then incubated with an MGMT mouse monoclonal antibody for one hour (NeoMarkers, Fremont, California, USA; dilution 1 in 50). Biotinylated rabbit antimouse immunoglobulin diluted 1 in 400 in 3% normal human serum was used as the secondary antibody. After washing, slides were incubated in peroxidase conjugated streptavidin-biotin complex (Dako) for 30 minutes. The peroxidase was demonstrated by incubating in diaminobenzidine solution for five minutes. After rinsing in water, nuclei were lightly counterstained with haematoxylin for two minutes and coverslipped. Sections were then scored following discussion with and verification by a histopathologist (Andrew Hanby) for the presence, location, and intensity of staining (2, strong/moderate ( = normal); 1, weak ( = reduced); 0, no staining ( = undetectable)). Normal epithelium and stromal cells provided a positive internal control on each section and were graded as having strong/moderate staining.
For western blotting, colon cancer cell line pellets were lysed in 0.1 M DTT/bromophenol blue and separated on a 12.5% resolving polyacylamide gel. After transfer to PVDF membranes (Millipore, Milton Keynes, UK) and blocking in 5% bovine serum albumin (Sigma, Poole, UK), the primary antibodies were exposed to the membrane overnight at 4°C with gentle agitation. The anti-MGMT mouse monoclonal antibody (Ab-1; NeoMarkers) was diluted 1 in 500, and the control anti-beta-actin mouse monoclonal antibody (Sigma) was diluted 1 in 5000 and exposed to the membrane both simultaneously and separately. After detection of the proteins with ECL reagents (Amersham, UK) according to the manufacturer’s instructions, the membrane was exposed to film for one and five minutes. MGMT protein levels were measured and normalised to beta-actin expression using NIH-image software (http://rsb.info.nih.gov/nih-image/Default.html).
RESULTS
In our series of frozen colorectal cancers, we found four missense MGMT mutations which were not known polymorphisms (table 1 ▶). In none of these cases was the variant present in the constitutional DNA. Tumour Nos 684 and 1350 had the missense mutation A75D, tumour No 6 had acquired a K104E change, and tumour No 930 had the missense variant G23D. Tumour No 6 showed LOH at MGMT and sequence inspection suggested that this involved the wild-type allele. In our panel of colorectal cell lines, we found two heterozygous missense mutations, G55C in DLD1 and G156C in SW48; sequence inspection provided no evidence for loss of the wild-type allele in either case. Although constitutional DNA did not exist for these two cell lines, we did not find G55C or G156C changes in any of the constitutional DNAs from the frozen cancer set, in a set of 96 UK control individuals or in any SNP database. The previously described MGMT polymorphisms K178R, I143V, L84F, and L53L were also found in samples of all types. No silent MGMT mutations were found.
Table 1.
Summary of changes in cancers with O6-methylguanine methyltransferase (MGMT) mutations
| Cancer | MSI status | K-ras mutation | p53 mutation | MGMT mutation | LOH | MGMT mRNA | MGMT protein |
| 930 | High | None | None | G23D G:C>A:T | No | N/D | Undetectable |
| DLD1 | High | G12C G:C>T:A | S241F G:C>A:T | G55C G:C>T:A | No | Mutant only | Normal |
| 684 | Stable | None | Normal | A75D G:C>T:A | No | N/D | Undetectable |
| 1350 | Low | None | Normal | A75D G:C>T:A | No | N/D | Normal |
| 6 | Low | None | R248W G:C>A:T | K104E A:T>G:C | Yes | N/D | Undetectable |
| SW48 | High | None | R248W G:C>A:T | G156C G:C>T:A | No | Mutant only | Reduced |
MSI, level microsatellite instability; LOH, loss of heterozygosity.
A homology search showed that MGMT genes have moderate cross species sequence homology (fig 1 ▶) although topology and overall structure have been reported to be well conserved. A series of approximately 30 MGMT residues in the carboxyl half of the protein is however absolutely or highly conserved; this region includes the DNA binding domain and the cysteine acceptor site which comprises the consensus sequence V(1)PCHRV(1). G23D occurred at a site that is fully conserved throughout higher organisms and specie, although its function is unclear. G55C occurred at a highly conserved site within a stretch of residues E45-G55 which has been reported to form a hydrophilic handle, which may serve to anchor MGMT to other proteins. We found A75D to be at a residue conserved across the mammalian species but this amino acid lies in a region of the protein with no known functional significance. K104E occurred in the MGMT DNA binding domain (codons 92–175). Residue 104 is highly conserved and lies within a sequence, LWKLLKVVKFGG, which is invariant in mammals (fig 1 ▶). G156C occurred at a highly conserved site just outside the cysteine acceptor domain. It has been shown27,28 that all substitutions at G156 confer resistance to the toxic MeG homologue O6-benzylguanine through protein backbone distortions which disturb the adjacent wall of the benzyl binding pocket, and the amount of bacterial protein required to obtain 80% repair of methylated DNA substrate is increased by eight times over the wild-type.
Figure 1.
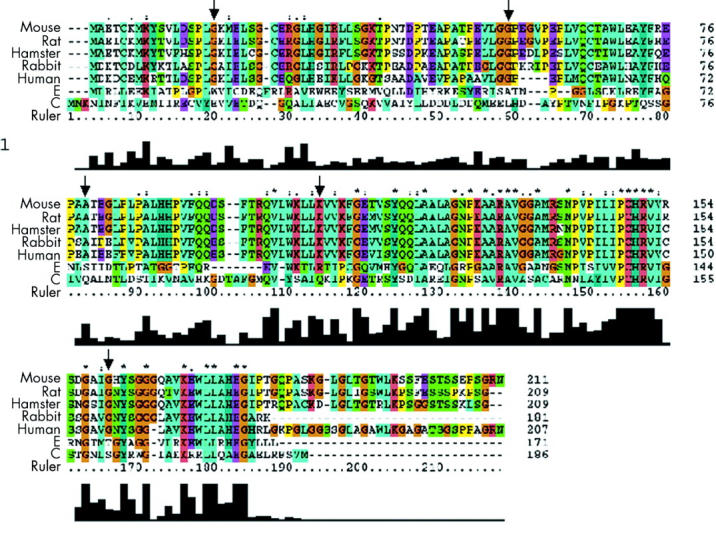
Alignment of O6-methylguanine methyltransferase (MGMT) protein across seven species. Positions of the mutations detected in this study are arrowed.
We determined mRNA and protein expression in the cell lines, DLD1 and SW48, which harboured missense mutations (table 1 ▶). Both lines expressed only mutant transcripts. MGMT protein was present at a level which was not notably reduced in DLD1 but protein levels were low in SW48. Although we could not determine mRNA expression in the four fresh frozen cancers, we analysed MGMT protein expression using immunohistochemistry. Tumour Nos 6, 684, and 930 had undetectable MGMT expression but tumour No 1350 had retained expression. There appeared prima facie to be a tendency to G:C>A:T changes at MGMT itself, K-ras, or p53 in cancers with MGMT mutations although no association with MSI status was evident (table 1 ▶).
MGMT immunohistochemistry on the full set of fresh frozen cancers showed that 19/63 (30%) tumours retained expression, eight (13%) had reduced expression, and 37 (58%) had undetectable expression. Of 81 tumours informative at microsatellite markers around the MGMT locus, 49 (60%) showed LOH. There was no association between LOH and reduced/undetectable MGMT protein expression (p>0.3, Fisher’s exact test).
Expression of MGMT protein was reduced or absent in 15 of 31 (47%) colorectal cancer cell lines analysed; a bimodal distribution of expression levels was evident (figs 2 ▶, 3 ▶). Reduced or normal protein expression in the cell lines segregated perfectly (details not shown) with promoter methylation which had been previously reported in the same lines.1,16,29
Figure 2.

O6-methylguanine methyltransferase (MGMT) protein expression in some of the colorectal cancer cell lines.
Figure 3.

Bimodal distribution of O6-methylguanine methyltransferase (MGMT) protein expression in colorectal cancer cell lines, showing clear evidence of group with reduced expression.
We assessed associations between MGMT expression in the full set of fresh frozen cancers and cancer cell lines and (i) presence of mutations in APC, beta-catenin, K-ras, and p53 and (ii) G:C>A:T changes at each of these loci. No association was found between reduced/absent MGMT expression and presence or absence of any mutation; details for K-ras are shown in table 2 ▶. In addition, there were no associations between absent/decreased MGMT expression and G:C>A:T mutations at any one genetic locus (details for K-ras and p53 in tables 3 ▶ and 4 ▶). However, considering all four genes together, the presence of one or more G:C>A:T changes was weakly associated with reduced or absent MGMT compared with cancers which had other types of mutation in these genes (table 5 ▶). MGMT expression was not associated with the MSI low phenotype (table 6 ▶) or with CIN status (details not shown), irrespective of whether or not MSI-high cancers were included in the analysis.
Table 2.
O6-methylguanine methyltransferase (MGMT) expression and K-ras mutation in the whole sample set
| MGMT expression | Total | ||
| Undetectable/reduced | Normal | ||
| K-ras mutation | |||
| Absent | 40 | 17 | 57 |
| Present | 19 | 18 | 37 |
| Total | 59 | 35 | 94 |
Undetectable/reduced expression was assessed using immunohistochemistry for frozen tumours and western analysis for cancer cell lines.
For this analysis, p = 0.05 (Fisher’s exact test). Note that the borderline association is actually in the opposite direction to that detected by Esteller and colleagues.20
Table 3.
O6-methylguanine methyltransferase (MGMT) expression and G:C>A:T K-ras mutations in the whole sample set
| MGMT expression | Total | ||
| Undetectable/reduced | Normal | ||
| K-ras G:C>A:T | 11 | 9 | 20 |
| Other K-ras | 9 | 9 | 18 |
| Total | 20 | 18 | 38 |
For this analysis, p = 0.51 (Fisher’s exact test).
Table 4.
O6-methylguanine methyltransferase (MGMT) expression and G:C>A:T p53 mutations in the whole sample set
| MGMT expression | Total | ||
| Undetectable/reduced | Normal | ||
| P53 G:C>A:T | 11 | 2 | 13 |
| Other p53 | 11 | 5 | 16 |
| Total | 22 | 7 | 29 |
For this analysis, p = 0.29 (Fisher’s exact test).
Table 5.
O6-methylguanine methyltransferase (MGMT) expression and any G:C>A:T mutation in the whole sample set
| MGMT expression | Total | ||
| Undetectable/reduced | Normal | ||
| Any G:C>A:T | 30 | 13 | 43 |
| No G:C>A:T | 10 | 12 | 22 |
| Total | 40 | 25 | 65 |
For this analysis, p = 0.05 (Fisher’s exact test).
Table 6.
O6-methylguanine methyltransferase (MGMT) expression and low level microsatellite instability (MSI low) in fresh frozen cancers, excluding MSI high tumours
| MGMT expression | Total | ||
| Undetectable/reduced | Normal | ||
| MSI low | 29 | 13 | 42 |
| Non-MSI low | 15 | 2 | 17 |
| Total | 44 | 15 | 59 |
Note that MSI low cannot be scored in the cell lines owing to lack of constitutional DNA.
For this analysis, p = 0.11 (Fisher’s exact test).
DISCUSSION
We have found six MGMT mutations in a series of 81 sporadic colorectal cancers and 32 colorectal cancer cell lines. All mutations were missense changes which were definitely or almost certainly not present in the germline. Unequivocal demonstration of the functional effects of each mutation is difficult but we have gained clues from homology searches, reported functional studies in lower organisms, and mutation spectra in the cancers themselves. We suspect that the mutations may greatly diminish, rather than abolish, MGMT function: we found no protein truncating changes and it is notable that studies looking at the effects of multiple amino acid substitutions have shown that only a base substitution of the cysteine 145 methyl acceptor site renders the protein entirely inactive.30
It is likely that K104E (tumour No 6) adversely affects MGMT function on the basis of its location within the highly conserved DNA binding domain of the protein (fig 1 ▶). This tumour showed loss of the wild-type MGMT allele, a G:C>A:T mutation in p53, and undetectable MGMT protein expression. G156C (SW48) occurred at a site previously shown to diminish MGMT function when mutated.28 This cell line also showed a G:C>A:T mutation in p53, loss of expression of the wild-type MGMT allele, and reduced expression of MGMT protein. DLD1 only expressed the mutant G55C mRNA (even though protein levels were normal) and harboured G:C>T:A changes at both K-ras and p53. Previous studies have reported DLD1 to be methylated at MGMT31 and, given that the mutant allele was expressed (and is therefore presumably unmethylated), it is probable that G55C reduces MGMT function.
The functional consequences of the other mutations, G23D and A75D, remain uncertain. Although these residues are highly conserved across species, they do not lie in regions of known functional importance within the protein. A75D was associated with normal protein levels in cancer No 1350, but undetectable protein in cancer No 684, suggesting silencing of that allele independent of the mutation in the latter tumour. G23D was associated with undetectable protein in cancer No 930. Neither A75D nor G23D was associated with G:C>A:T mutations in APC, K-ras, or p53.
Although we found no association between MGMT protein expression and the presence or type (G:C>A:T) of mutation at APC, beta-catenin, K-ras, or p53 when analysed separately, the presence of any G:C>A:T mutation at one or more of these loci was associated, albeit weakly, with reduced MGMT expression. Reduced MGMT expression was not however associated with an increased frequency of K-ras mutations (of any type) or with the MSI low phenotype, thus supporting the data of Laiho and colleagues.21
Analysis of associations between MGMT changes and mutation spectra is intrinsically difficult. It is, for example, impossible to reconstruct the mutagenic lesion which led to an observed mutation, and G>A changes are usually indistinguishable from the more common C>T mutations caused by spontaneous deamination of (5-methyl) cytosine. For understandable reasons, the full extent of MGMT methylation has rarely been determined in cancer studies, and the effects of partial or hemi methylation are unclear. Some doubt has existed therefore about the importance of reduced MGMT expression in colorectal carcinomas and the validity of its association with G:C>A:T changes. It has therefore been reasonable to suggest that methylation of the MGMT promoter is a background event which does not promote tumorigenesis. The finding of somatic MGMT mutations with probable functional consequences provides good evidence to show that decreased MGMT function is selected in some colorectal carcinomas. Our data imply that methylation of MGMT is, at least sometimes, not a background event but a selected change with functional consequences. As MGMT provides only one of several lines of defence against G:C>A:T mutations, the effect of reduced MGMT expression on mutation spectra is probably a relatively subtle bias towards these changes.32
Acknowledgments
We are grateful to Andrew Hanby (Department of Histopathology, Leeds) for assistance with immunohistochemistry scoring. The Equipment Park of the London Research Institute, Cancer Research UK provided invaluable support.
Abbreviations
MGMT, O6-methylguanine methyltransferase
MeG, O6-methylguanine
MSI low, low level microsatellite instability
PCR, polymerase chain reaction
CIN, chromosomal instability
F-SSCP, fluorescent-single strand conformational polymorphism
LOH, loss of heterozygosity
Conflict of interest: None declared.
REFERENCES
- 1.Aquilina G, Biondo R, Dogliotti E, et al. Expression of the endogenous O6-methylguanine-DNA-methyltransferase protects Chinese hamster ovary cells from spontaneous G:C to A:T transitions. Cancer Res 1992;52:6471–5. [PubMed] [Google Scholar]
- 2.Pegg AE, Dolan ME, Moschel RC. Structure, function, and inhibition of O6-alkylguanine-DNA alkyltransferase. Prog Nucleic Acid Res Mol Biol 1995;51:167–223. [DOI] [PubMed] [Google Scholar]
- 3.Zaidi NH, Pretlow TP, O’Riordan MA, et al. Transgenic expression of human MGMT protects against azoxymethane-induced aberrant crypt foci and G to A mutations in the K-ras oncogene of mouse colon. Carcinogenesis 1995;16:451–6. [DOI] [PubMed] [Google Scholar]
- 4.Hampson R, Humbert O, Macpherson P, et al. Mismatch repair defects and O6-methylguanine-DNA methyltransferase expression in acquired resistance to methylating agents in human cells. J Biol Chem 1997;272:28596–606. [DOI] [PubMed] [Google Scholar]
- 5.Costello JF, Futscher BW, Kroes RA, et al. Methylation-related chromatin structure is associated with exclusion of transcription factors from and suppressed expression of the O-6-methylguanine DNA methyltransferase gene in human glioma cell lines. Mol Cell Biol 1994;14:6515–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris LC, Remack JS, Brent TP. In vitro methylation of the human O6-methylguanine-DNA methyltransferase promoter reduces transcription. Biochim Biophys Acta 1994;1217:141–6. [DOI] [PubMed] [Google Scholar]
- 7.von Wronski MA, Brent TP. Effect of 5-azacytidine on expression of the human DNA repair enzyme O6-methylguanine-DNA methyltransferase. Carcinogenesis 1994;15:577–82. [DOI] [PubMed] [Google Scholar]
- 8.Qian X, von Wronski MA, Brent TP. Localization of methylation sites in the human O6-methylguanine-DNA methyltransferase promoter: correlation with gene suppression. Carcinogenesis 1995;16:1385–90. [DOI] [PubMed] [Google Scholar]
- 9.Skorpen F, Krokan HE. The methylation status of the gene for O6-methylguanine-DNA methyltransferase in human Mer+ and Mer− cells. Carcinogenesis 1995;16:1857–63. [DOI] [PubMed] [Google Scholar]
- 10.Patel SA, Graunke DM, Pieper RO. Aberrant silencing of the CpG island-containing human O6-methylguanine DNA methyltransferase gene is associated with the loss of nucleosome-like positioning. Mol Cell Biol 1997;17:5813–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian XC, Brent TP. Methylation hot spots in the 5′ flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res 1997;57:3672–7. [PubMed] [Google Scholar]
- 12.Kokkinakis DM, Ahmed MM, Delgado R, et al. Role of O6-methylguanine-DNA methyltransferase in the resistance of pancreatic tumors to DNA alkylating agents. Cancer Res 1997;57:5360–8. [PubMed] [Google Scholar]
- 13.Watts GS, Pieper RO, Costello JF, et al. Methylation of discrete regions of the O6-methylguanine DNA methyltransferase (MGMT) CpG island is associated with heterochromatinization of the MGMT transcription start site and silencing of the gene. Mol Cell Biol 1997;17:5612–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao W, Samson L. In vivo evidence for endogenous DNA alkylation damage as a source of spontaneous mutation in eukaryotic cells. Proc Natl Acad Sci U S A 1993;90:2117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson PE, Hall CN, O’Connor PJ, et al. Low O6-alkylguanine DNA-alkyltransferase activity in normal colorectal tissue is associated with colorectal tumours containing a GC-->AT transition in the K-ras oncogene. Carcinogenesis 1997;18:1299–302. [DOI] [PubMed] [Google Scholar]
- 16.Esteller M, Hamilton SR, Burger PC, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 1999;59:793–7. [PubMed] [Google Scholar]
- 17.Esteller M, Fraga MF, Guo M, et al. DNA methylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet 2001;10:3001–7. [DOI] [PubMed] [Google Scholar]
- 18.Esteller M, Risques RA, Toyota M, et al. Promoter hypermethylation of the DNA repair gene O(6)-methylguanine-DNA methyltransferase is associated with the presence of G:C to A:T transition mutations in p53 in human colorectal tumorigenesis. Cancer Res 2001;61:4689–92. [PubMed] [Google Scholar]
- 19.Whitehall VL, Walsh MD, Young J, et al. Methylation of O-6-methylguanine DNA methyltransferase characterizes a subset of colorectal cancer with low-level DNA microsatellite instability. Cancer Res 2001;61:827–30. [PubMed] [Google Scholar]
- 20.Esteller M, Toyota M, Sanchez-Cespedes M, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is associated with G to A mutations in K-ras in colorectal tumorigenesis. Cancer Res 2000;60:2368–71. [PubMed] [Google Scholar]
- 21.Laiho P, Launonen V, Lahermo P, et al. Low-level microsatellite instability in most colorectal carcinomas. Cancer Res 2002;62:1166–70. [PubMed] [Google Scholar]
- 22.Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamashita K, Dai T, Dai Y, et al. Genetics supersedes epigenetics in colon cancer phenotype. Cancer Cell 2003;4:121–31. [DOI] [PubMed] [Google Scholar]
- 24.Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci U S A 2000;97:3352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halford SE, Sawyer EJ, Lambros MB, et al. MSI-low, a real phenomenon which varies in frequency among cancer types. J Pathol 2003;201:389–94. [DOI] [PubMed] [Google Scholar]
- 26.Sieber OM, Heinimann K, Gorman P, et al. Analysis of chromosomal instability in human colorectal adenomas with two mutational hits at APC. Proc Natl Acad Sci U S A 2002;99:16910–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu-Welliver M, Kanugula S, Loktionova NA, et al. Conserved residue lysine165 is essential for the ability of O6-alkylguanine-DNA alkyltransferase to react with O6-benzylguanine. Biochem J 2000;347 (Pt 2) :527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu-Welliver M, Pegg AE. Point mutations at multiple sites including highly conserved amino acids maintain activity, but render O6-alkylguanine-DNA alkyltransferase insensitive to O6-benzylguanine. Biochem J 2000;347 (Pt 2) :519–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lind GE, Thorstensen L, Lovig T, et al. A CpG island hypermethylation profile of primary colorectal carcinomas and colon cancer cell lines. Mol Cancer 2004;3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chueh LL, Nakamura T, Nakatsu Y, et al. Specific amino acid sequences required for O6-methylguanine-DNA methyltransferase activity: analyses of three residues at or near the methyl acceptor site. Carcinogenesis 1992;13:837–43. [DOI] [PubMed] [Google Scholar]
- 31.Branch P, Hampson R, Karran P. DNA mismatch binding defects, DNA damage tolerance, and mutator phenotypes in human colorectal carcinoma cell lines. Cancer Res 1995;55:2304–9. [PubMed] [Google Scholar]
- 32.Sandercock LE, Kwok MC, Luchman HA, et al. Mutational-reporter transgenes rescued from mice lacking either Mgmt, or both Mgmt and Msh6 suggest that O6-alkylguanine-induced miscoding does not contribute to the spontaneous mutational spectrum. Oncogene 2004;23:5931–40. [DOI] [PubMed] [Google Scholar]