

Prompt removal of unwanted cells, such as senescent, damaged, genetically mutated, or virus infected cells, is crucial for the maintenance of liver health. This process is naturally achieved through a highly regulated programmed form of cell death called apoptosis. In healthy organisms, the number of cells eliminated by apoptosis equals the number of cells generated by mitosis, ensuring the proper organ homeostasis. In addition, “physiological” apoptosis allows the removal of cells with virtually no release of proinflammatory cytokines and minimal immune response. However, in pathophysiological situations, the balance between cell proliferation and cell death is often altered, with the consequent loss of tissue homeostasis and the onset of several liver diseases. Insufficient apoptosis, with failure of removal of cells carrying mutated genes, and unregulated proliferation within the context of a persistent inflammatory milieu, can promote the development of liver and biliary cancer.1–4 Paradoxically, a chronic apoptotic stimulus can also predispose to cancer development due to the high rate of regeneration invoked in the tissue, which elevates the risk of mitotic errors. In contrast, excessive and/or sustained apoptosis can lead to acute injuries, such as fulminant hepatitis and reperfusion damage,5,6 or even chronic sustained injuries, such as alcoholic liver disease, cholestatic liver disease, and viral hepatitis.7–10 Therefore, therapeutic strategies to inhibit apoptosis in liver injury, or selectively kill malignant cells in tumours, have the potential to provide a powerful tool for the treatment of liver disease. Indeed, with an improved understanding of the molecular pathways and the pathophysiological role of apoptosis, new drugs aimed at therapeutically modulating apoptosis are now available for clinical trials and/or as new therapeutic options for the treatment of several human diseases. In this review, we will focus on the role of apoptosis in selected liver diseases, such as alcoholic liver disease, viral hepatitis, cholestatic liver diseases, non-alcoholic liver disease, and hepatocellular carcinoma.11–14 We will also review some pro- and antiapoptotic therapies currently in use (or in clinical trial) or potentially useful for the treatment of human diseases, including liver diseases.
DEFINITION OF APOPTOSIS
Apoptosis is a highly organised and genetically controlled type of cell death, essential during embryonic development to ensure proper organogenesis15 as well as for the health of adult organisms. It is characterised by a number of distinct morphological alterations, such as chromatin condensation and marginalisation, cell shrinkage, and plasma membrane blebbing, which are accompanied by biochemical features such as DNA fragmentation, membrane alterations (that is, exposure of phosphatidylserine on the outside of the plasma membrane), and degradation of specific cellular proteins, as a result of the massive activation of a large number of intracellular proteases and endonucleases. In the latest stages, the dying cell is fragmented into membrane bound vesicles containing relatively intact organelles and chromatin residues named “apoptotic bodies”16 which are readily engulfed by neighbouring cells and professional phagocytes, such as resident macrophages or, in the liver, by Kupffer cells (fig 1 ▶). Supposedly, the efficient clearance of dead cells prevents triggering of the immune response that may follow spontaneous lysis of the apoptotic bodies and release of proinflammatory cytokines, making it ideal for remodelling of the tissue. Perhaps the most remarkable feature of apoptosis is its highly regulated nature. Apoptosis occurs through activation of a cell death machinery via specific molecular pathways that are under the tight control of a complex network of proteins and their endogenous inhibitors. This allows the cell to control its fate by starting the apoptotic programme and relieving the apoptotic inhibitions, whenever is necessary, or preventing the engagement of the death machinery under normal conditions. The control occurs mainly at checkpoints in the pathways that can also potentially be used as targets for therapeutic modulation of apoptosis.
Figure 1.

Evidence of hepatocyte apoptosis in vivo. Histopathological examination of a liver section from a patient with hepatitis C virus by conventional haematoxylin-eosin staining shows an apoptotic body (also known as Councilman body, black arrow) surrounded by immune cells, such as T lymphocytes (white arrows) and macrophages.
Many of the above concepts are however likely not relevant to “apoptosis” in pathobiology. Whereas physiological apoptosis is tightly restricted to discrete subsets of cells, both spatially and temporally, pathological apoptosis involves large numbers of cells, is non-selective, can be sustained over time, and often occurs in an inflammatory milieu. Therefore, the concept of apoptosis in disease must be re-evaluated. For example, we now know that single disruption of an antiapoptotic gene results in sustained serum ALT elevations,17 and specific apoptotic protease mediated cleavage products circulate in the serum of HCV infected individuals.18 Obviously, based on these observations, it is clear that sustained apoptosis is associated with release of cellular constituents into the extracellular space and serum. Lipid products from apoptotic cells can also serve as chemotactic factors and recruit inflammatory cells (fig 1 ▶).19 Apoptosis of liver cells has now been linked to liver fibrosis in several studies. Thus the physiological concept that liver cell apoptosis is “innocuous” cannot be transferred to pathological apoptosis.
▸ Under physiological conditions, apoptosis is a highly regulated form of cell death, crucial for the development and proper functioning of all multicellular organisms.
▸ Pathological apoptosis occurs in an unregulated fashion, and can be sustained and injurious.
▸ Several diseases, including many liver diseases, are associated with dysregulation of apoptosis.
▸ Therapeutic modulation of apoptosis may represent a valid strategy for the treatment of human liver diseases.
MECHANISMS OF APOPTOSIS
In order to develop new strategies aimed at modulating apoptosis in the treatment of liver disease, it is necessary to understand the molecular mechanisms that regulate the apoptotic process.
Although apoptosis can be triggered by several different stimuli, apoptotic signalling within the cell is transduced mainly via two defined molecular pathways: the death receptor pathway (also called the extrinsic pathway) and the mitochondrial pathway (also called the intrinsic pathway). The end point of both the intrinsic and extrinsic pathways is activation of a wide variety of intracellular proteases (especially a group of proteolytic enzymes called caspases) and endonucleases that ultimately degrade the cellular constituents. The extrinsic pathway originates at the plasma membrane following the engagement of a family of cytokine receptors named death receptors (such as tumour necrosis factor receptor 1 (TNF-R1), Fas/CD95, and tumour necrosis factor related apoptosis inducing ligand receptors 1 and 2 (TRAIL-R1 and TRAIL-R2)) by their cognate ligands (TNF-α, Fas ligand (FasL)/CD95L, TRAIL).20 Ligand/receptor binding induces the recruitment of several adapter proteins and proenzymes (that is, procaspase-8 and -10) at the intracellular domain of the receptor to form a complex usually referred to as DISC (death inducing signalling complex). The signal generated at the DISC by activated caspases results in cell death which, depending on the cell type, may or may not require the involvement of mitochondria for its execution (fig 2 ▶). The intrinsic pathway is triggered by different extra- or intracellular signals, such as γ irradiations, oxidative stress, toxins, reactive intermediates of xenobiotic metabolism, endoplasmic reticulum stress inducing factors, growth factor deprivation, or some chemotherapeutic drugs which induce mitochondrial dysfunction.21,22 As a result, organelle architecture and membrane permeability are altered, and mitochondrial proteins are released into the cytosol, including proapoptogenic factors such as cytochrome c, SMAC/DIABLO (second mitochondria derived activator of caspases/direct IAP binding protein with low pI), HtrA2/Omi, apoptosis inducing factor, and endonuclease G (fig 3 ▶), which contribute to protease activation and chromatin degradation. The extrinsic and intrinsic pathways are not mutually exclusive, as some cells, including hepatocytes and cholangiocytes, have been shown to require mitochondrial involvement to amplify the apoptotic signal from death receptors.
Figure 2.
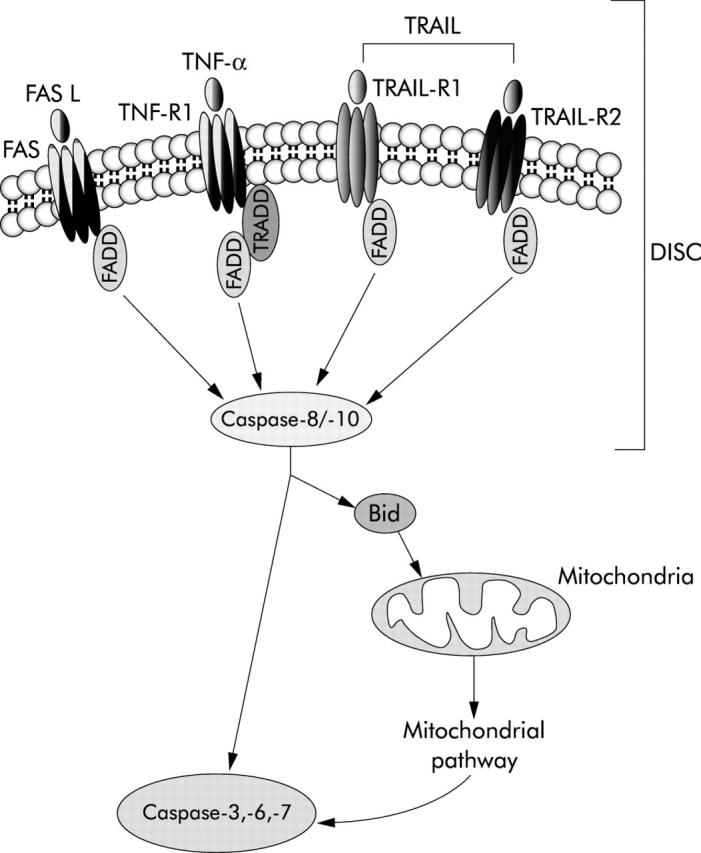
Death receptor mediated (extrinsic) pathway of apoptosis. Schematic representation of signalling through the main death receptors (Fas/CD95, tumour necrosis factor receptor 1 (TNF-R1), tumour necrosis factor related apoptosis inducing ligand receptors 1 and 2 (TRAIL-R1 and TRAIL-R2)). Engagement of death receptors by their cognate ligands results in oligomerisation of the receptor and recruitment of adaptor proteins (Fas associated protein with death domain (FADD), TNF-R1 associated death domain protein (TRADD)), which in turn bind the inactive initiator caspase-8 and/or -10. The resulting complex is referred to as the death inducing signalling complex (DISC). The close proximity of several procaspase molecules results in activation of the caspase by self processing. Active initiator caspases can directly activate downstream caspases such as caspase-3, -6, and -7, or engage the mitochondrial pathway of apoptosis by cleavage and activation of the BH3 only protein Bid (see fig 3 ▶ for details).
Figure 3.

Mitochondria mediated (intrinsic) pathway of apoptosis. Various stimuli, including ultraviolet (UV) and γ-irradiation, endoplasmic reticulum (ER) stress, growth factor deprivation, and oxidative stress with production of reactive oxygen species (ROS) trigger the intrinsic pathway via activation of proapoptotic members of the Bcl-2 family of protein (that is, Bax, Bak), which oligomerise on the outer mitochondrial membrane and cause mitochondrial dysfunction. The proapoptotic action of Bax and Bak can be antagonised by the antiapoptotic members of the same family Bcl-2 or Bcl-XL. Following mitochondrial dysfunction, several apoptogenic factors, including cytochrome c and second mitochondria derived activator of caspases/direct IAP binding protein with low pI (SMAC/DIABLO), are released from the mitochondrial intermembrane space into the cytosol. Cytochrome c binds to the adaptor apoptosis associated factor 1 (Apaf-1) and recruits procaspase-9 to form a complex named apoptosome which, in an APT requiring reaction, results in activation of the initiator caspase-9. Caspase-9, in turn, activates the effector caspases (caspase-3, -6, and -7) responsible for degradation of cellular substrates. SMAC/DIABLO contributes to caspase activation by binding and inactivating the endogenous inhibitor of caspases IAPs.
A group of proteases, in particular the caspases (cysteinyl aspartate specific proteases), play a central role as executors of the cell death programme.23 Caspases are constitutively expressed as inactive proenzymes and generally require proteolytic processing for their activation. As caspases cleave substrates on the carboxy terminal side of an Asp residue, and they also require cleavage at Asp sites to acquire their catalytic activity, caspases are capable of self activation, as well as of activating each other in a cascade-like process. Among the 14 mammalian caspases identified to date, 12 of which have also been cloned in humans, some are primarily involved in apoptosis (caspases-2, -3, -6, -7, -8, -9, -10, and -12). These caspases can be divided into either upstream caspases (also known as initiator or apical caspases) or downstream caspases (also called effector or distal caspases). Upstream caspases (-2, -8, -9, -10) are activated following binding to adaptors (such as FADD or apoptosis associated factor 1), which promotes self association and autocatalytic activation. In contrast, downstream caspases (-3, -6, and -7) lack the ability to self associate and require cleavage by initiator caspases for their activation. Activated downstream caspases are responsible for degradation of several cellular substrates associated with the morphological changes of apoptosis, including nuclear degradation, cytoskeleton alterations, and membrane blebbing. Other caspases, such as caspases-1, -4, -5, and -11, are involved in inflammation.24 This concept is important to note in that systemic administration of pan-caspase inhibitors also disrupts many inflammatory cascades. Therefore, the use of these agents cannot be solely ascribed to blocking apoptosis. The ability to both disrupt inflammation and apoptosis however may be therapeutically very beneficial.
Several intracellular proteins are involved in the regulation of apoptosis. In particular, the Bcl-2 family of proteins, which includes both pro- and antiapoptotic members, are perhaps the most important regulators of the intrinsic pathway. These proteins act upstream and at the level of the mitochondria to integrate death and survival signals, and the balance between pro- and antiapoptotic members of the family, as well as their reciprocal interactions, determines whether or not the intrinsic pathway of apoptosis is initiated. To date, the mammalian Bcl-2 family comprises at least 20 members with various degrees of homology within four conserved regions, named Bcl-2 homology (BH) 1–4 domains.25,26 The family can be further divided into three main subclasses, defined in part by this homology and in part by their function. The first subclass includes, among others, the antiapoptotic proteins Bcl-2, Bcl-XL, and Mcl-1, which share the highest homology in all of the four conserved BH 1–4 domains. Their localisation is generally mitochondrial but some have also been found on the endoplasmic reticulum and nuclear membrane. The mechanisms by which they prevent mitochondrial dysfunction are still largely unclear although they have been found to directly bind to several proapoptotic members of the family.27 The second and third subclasses include only proapoptotic proteins: the so-called multidomain Bax-like proteins, such as Bax and Bak, and the BH3 only proteins, such as Bid, Bad, Bim, Noxa, and Puma. Both multidomain and BH3 only proteins are required for apoptosis. In healthy cells, Bax is found in the cytosol as a monomer, but following an apoptotic stimulus it undergoes conformation changes, integrates into the outer mitochondrial membrane, and oligomerises, causing membrane permeabilisation28–30 Bak is an oligomeric integral mitochondrial membrane protein but it also undergoes conformational changes during apoptosis and forms larger aggregates.30,31 Following specific death signals, BH3 only proteins are activated which in turn activate Bax or Bak either by directly interacting with them32 or by binding to and antagonising the antiapoptotic members of the family.33 Activation of either Bax or Bak triggers mitochondrial dysfunction and is required for apoptosis, but the mechanism(s) through which mitochondrial permeabilisation is achieved is still debated (fig 3 ▶).34 The BH3 only protein Bid also provides crosstalk between the extrinsic and intrinsic pathways. Indeed, Bid is activated by caspase-8 following death receptor engagement and translocates to the mitochondria where it contributes to activation of Bax or Bak, and to mitochondrial dysfunction (fig 2 ▶).31,35,36
▸ Apoptosis occurs mainly via a two signalling pathways: a death receptor mediated extrinsic pathway or a mitochondria mediated intrinsic pathway. The pathways are not mutually exclusive.
▸ The cysteine proteases caspases are key executors of the apoptotic programme and are activated by both the extrinsic and intrinsic pathways; downstream caspases are directly responsible for degradation of several cellular components.
▸ Proteins of the Bcl-2 family are the main regulators of the intrinsic pathway; they serve as sensors to integrate death and survival signals at the level of the mitochondria, and the balance between pro- and antiapoptotic members of the family, as well as their interactions, determine whether mitochondria are permeabilised.
APOPTOSIS AND LIVER DISEASE
Death receptors, especially Fas, are widely expressed in all liver cell types, likely in response to the evolutionary pressure to eliminate hepatotropic viruses.37 The Fas/FasL system is indeed the pathway most commonly used by immunocytes to kill virally infected cells.38 Because of this high level of death receptor expression in hepatocytes, apoptosis in the liver occurs mainly via the extrinsic pathway (table 1 ▶). However, in both hepatocytes and cholangiocytes, mitochondria are also engaged by the death receptor pathway via activation of Bid,39,40 and therefore alterations in the intrinsic pathway are often reported in liver diseases. Based on the above concepts, we will now explore what is known about apoptotic pathways in acute and chronic liver injuries.
Table 1.
Fas/Fas ligand mediated apoptosis in human liver diseases
| Liver disease | Reference |
| Hepatocarcinoma | Nagao M, et al 199958 |
| Ito Y, et al 2000116 | |
| Lee SH, et al 2001117 | |
| Fukuzawa Y, et al 200159 | |
| Okano H, et al 2003118 | |
| Chronic viral hepatitis | Fiore G, et al 1999119 |
| Tagashira M, et al 2000120 | |
| Ehrmann J Jr, et al 2000121 | |
| Pianko S, et al 2001122 | |
| Bantel H, et al 2001123 | |
| Ibuki N, et al 2002124 | |
| Tang TJ, et al 2003125 | |
| Acute viral hepatitis | Rivero M, et al al 2002126 |
| Fulminant hepatic failure | Ryo K, et al 2000127 |
| Alcoholic hepatitis | Natori S, et al 20017 |
| Ziol M, et al 200181 | |
| Tagami A, et al 200386 | |
| Autoimmune hepatitis | Fox CK, et al 2001128 |
| HCV related fibrosis | Pianko S, et al 2001122 |
| Bantel H, et al 200418 | |
| Wilson’s disease | Strand S, et al 1998129 |
| Acute allograft rejection | Tannapfel A, et al 1999130 |
| Non-alcoholic steatohepatitis | Feldstein A, et al 200394 |
| Ribeiro PS, et al 200493 |
Hepatocellular carcinoma
Hepatocellular carcinoma (HCC) is the most common primary malignant tumour of the liver, with a vast incidence throughout the world. Its pathogenesis is multifactorial, with a strong aetiological association with chronic viral hepatitis, alcohol consumption, exposure to hepatic toxins, as well as some genetic disorders such as haemochromatosis and α1 antitrypsin deficiency. Especially in its promotion stage, HCC has been associated with defective apoptosis and increased cell proliferation. In particular, tumour cells often show alterations in expression of tumour suppressor genes, DNA repair genes, genes regulating the cell cycle, and genes involved in apoptosis.41
Among the most common alterations frequently observed in HCC, as well in numerous other tumours, are mutations of p53.42,43 The protein p53 is the product of a tumour suppressor gene activated as a result of DNA damage. To allow the repair of the DNA damage, p53 initially induces cell cycle arrest. However, if the damage is too extensive to be repaired and the cell has to be removed, p53 can also induce apoptosis by transcriptional upregulation of BH3 only proteins such as Noxa, Puma, and Bid, and/or the multidomain protein Bax. p53 can also promote apoptosis by upregulation of death receptors and death ligands, including TRAIL-R1, Fas, and FasL.44–46 In addition, a transcriptional independent mechanism for p53 mediated apoptosis has been described where it directly associates with and causes mitochondrial dysfunction.47 Therefore, p53 provide an effective mechanism to protect the organism from the accumulation and propagation of genetic lesions. A dysfunctional p53 allows the tumour cell to escape apoptosis and results in cancer development. In addition, as several chemotherapeutic drugs induce apoptosis of tumour cells by causing DNA damage and activation of p53, tumours with disrupted p53 are generally resistant to chemotherapy and associated with an unfavourable prognosis. Studies in vitro and in vivo have demonstrated that adenoviral mediated expression of wild-type p53 suppresses the transformed phenotype of many cell types and potentiates the cytotoxicity of both chemotherapeutic agents and radiation therapy.48 Several phase I and II studies have evaluated the safety, biological effect, and different routes of administration of adenoviral mediated p53 gene therapy in various tumour types, indicating that adenovirus mediated introduction of wild type p53 into tumour cells represents a potentially valuable tool for the therapy of many types of human cancers.49,50 However, only preliminary and not conclusive results are available to date from p53 gene therapy clinical trials for HCC.51
Another alteration leading to defective apoptosis that is frequently observed in several tumours, including HCC, is downregulation or loss of Fas expression.52,53 Loss of Fas, often accompanied by expression of FasL, represents an advantageous adaptation for cancer cells as it enables them not only to survive the attack carried by FasL expressing cytotoxic T lymphocytes and natural killer cells, but also to actively kill the immune cells and create immune privileged sites.54–56 Several studies have described complete or partial reduction of Fas expression in HCC, which negatively correlated with the degree of HCC differentiation and patient survival.55,57–59 For this reason, levels of Fas expression could also be used as a marker of dedifferentiation to predict HCC biological behaviour. However, as activated immune cells can induce apoptosis not only via engagement of the Fas/FasL pathway, but also through other death receptors, or via the perforin/granzyme pathway, downregulation of Fas alone may not be sufficient to escape the immune response. Indeed, many HCC have been found to also overexpress the antiapoptotic protein Bcl-XL, which confers resistance to mitochondria mediated apoptosis.17 As the death receptor mediated pathway of apoptosis is strictly linked to the mitochondrial pathway in hepatocytes, overexpression of Bcl-XL may contribute to Fas resistance in these tumours. Therapeutic approaches aimed at restoring Fas expression and sensitivity to Fas mediated apoptosis in tumour cells may therefore be proved useful in the therapy of HCC, as well as in other tumours. Several drugs currently in use for chemotherapy have been found to upregulate Fas expression via activation of p53 and increase sensitivity to Fas mediated apoptosis in tumour cells with wild-type p53.46,60 However, modulation of other component of the Fas/FasL apoptotic pathway, such as Bcl-XL, must be considered in designing new improved chemotherapeutic strategies.
Viral hepatitis
Infection by hepatitis B (HBV) or C virus (HCV), two of the seven human hepatitis viruses identified so far, is the main cause for viral hepatitis and represents a worldwide health problem. These viruses are able to persist in the host for years, contributing to the onset of chronic hepatitis. Moreover, because of continuous intrahepatic inflammation due to persistent infection, liver tissue undergoes a high rate of cell destruction and regeneration that results in an increased risk of developing HCC. HBV and HCV triggered liver injury is mediated mainly by host immune response to viral proteins expressed by infected hepatocytes and, to a lesser extent, by direct cytopathic effects of the virus. Indeed, during viral hepatitis, cytotoxic T lymphocytes recognise and kill viral antigen expressing HBV or HCV infected hepatocytes to clear the virus from the liver, causing the initial liver damage. Subsequently, the influx of antigen non-specific inflammatory cells exacerbates the tissue damage, with the formation of necroinflammatory foci. Several studies have documented that cytotoxic T lymphocytes kill virus infected hepatocytes by Fas dependent apoptosis, as demonstrated by enhanced Fas expression and increased number of FasL positive infiltrating mononuclear cells in the liver of hepatitis C patients,61–63 and patients with chronic active hepatitis B,64–66 which correlate with the severity and location of liver inflammation. Fas expression can be induced either by virus specific protein expression or by inflammatory cytokines such as interleukin 1, generated after the first immune response. Other pathways, including TNF-α and the perforin/granzyme system, have also been implicated in hepatocyte apoptotic processes in viral hepatitis.67,68 Caspase activation, triggered by death ligands, other cytokines, granzyme B, or HCV proteins, is considerably increased in HCV infected liver, and correlates closely with the inflammatory response.69 In this context, it is interesting to note that hepatocytes are resistant to granzyme B mediated cell death, and cytotoxic T lymphocytes kill virally infected hepatocytes almost exclusively by the Fas pathway.70 Finally, a recent study has demonstrated that patients with active HCV, independent of their alanine aminotransferase values, have elevated levels of caspase generated cytokeratin 18 cleavage fragments, a measurement of caspase activation in the liver, compared with healthy controls.18 These findings are of great clinical importance as they may have identified a new more sensitive biomarker for detecting apoptosis mediated liver injury that could be used both for disease diagnosis and as an end point for assessing therapies.
The role of the HBV X gene product (HBx) in hepatocyte apoptosis remains controversial. Studies with transgenic mice have demonstrated that HBx may stimulate the apoptotic turnover of hepatocytes.71 In contrast, HBx has also been reported to stimulate nuclear factor κB (NFκB) or JNK pathways, which block Fas induced apoptosis in liver cells.72,73 Similarly, single HCV proteins have been reported to have both pro- and antiapoptotic effects. Infection with the hepatitis C core protein increases susceptibility to Fas mediated apoptosis in a hepatoma cell line although the mechanism remains unclear and seems not to be due to increased Fas expression.74 In contrast, multiple HCV proteins (core, E1, E2, and NS2 proteins) expressed in transgenic mice have been shown to inhibit Fas mediated apoptosis by preventing the release of cytochrome c from the mitochondria and activation of caspase-9, -3, and -7.75 Therefore, hepatitis virus proteins may either sensitise hepatocytes to Fas induced apoptosis, critically contributing to liver damage, or inhibit apoptosis, as a possible mechanism to maintain persistent infection, and promote development of HCC.
In summary, the onset of hepatitis seems to proceed from an initial non-inflammatory event (apoptosis) to unspecific necroinflammation. The inflammatory process likely results from ineffective clearance of the apoptotic bodies by neighbouring phagocytes whose phagocytic capacity is overwhelmed by the large number of dying cells. The result is the release of potentially toxic or immunogenic intracellular contents, which elicits the inflammatory response and exacerbates tissue injury, leading to acute or fulminant hepatitis. In contrast, failure in eliminating infected hepatocytes as a result of virus elicited resistance may lead to viral persistence and promote the development of chronic hepatitis. Therefore, a therapeutic approach aimed at modulating immune cell mediated hepatocyte apoptosis may be appropriate to reduce liver damage during viral hepatitis. Preliminary studies recently showed effective reduction of liver damage and improvement of survival in mice injected with small interfering RNA (siRNA) against caspase-8 in models of acute liver failure (mediated by Fas agonistic agents) or acute viral hepatitis.76 Moreover, the same siRNA approach against Fas has been shown to protect mice from fulminant hepatitis, and prevent development of fibrosis in a model of chronic hepatitis.77 As a cautionary note, it has to be pointed out that although the data are solid and certainly promising, the applicability of this therapeutic approach to humans remains to be established. On the other hand, a nitric oxide derivative of ursodeoxycholic acid, NCX-1000, a compound that is selectively metabolised by hepatocytes, has been found to effectively protect against liver damage in murine models of autoimmune hepatitis induced by injection of concanavalin A or a Fas agonistic antibody, by inhibiting caspase activity. In particular, NCX-1000 protected against T helper 1 mediated liver injury by inhibiting both the proapoptotic and proinflammatory branches of the caspase superfamily, demonstrating that caspase inhibition may be a valid therapeutic strategy to reduce immune cell mediated liver damage.78 Finally, phase II trials are currently ongoing to explore the effect of a pan-caspase inhibitor in HCV patients unresponsive to approved antiviral agents.
Alcoholic hepatitis
The pathogenesis of alcoholic hepatitis and its degeneration to alcoholic cirrhosis are poorly understood. Data obtained from studies employing models of experimental ethanol induced liver injury have highlighted the crucial role of apoptosis in this type of liver damage.79,80 However, the importance of apoptosis in alcoholic liver diseases has only recently been demonstrated. Hepatocyte apoptosis has been observed in patients with alcoholic hepatitis, and directly correlates with disease severity, being most abundant in patients with high bilirubin and aspartate aminotransferase levels and grade 4 steatohepatitis.7,81 Apoptotic hepatocytes often colocalise with infiltrating neutrophils, suggesting an inflammatory response triggered by apoptosis.81–83 Among the several mechanisms proposed to explain alcohol induced hepatocyte apoptosis, induction of CYP2E1, one of the many cytochrome P450 isoforms, and CYP2E1 dependent formation of reactive oxygen species (ROS) and lipid peroxides, appears to be one of the possible explanations.84,85 ROS, whose production is driven by increased availability of the reduced form of nicotinamide adenine dinucleotide due to mitochondrial acetaldehyde metabolism, may cause mitochondrial dysfunction and release of proapoptotic factors such as cytochrome c into the cytosol where they promote caspase activation. Consistently, antioxidants have been shown to reduce hepatocyte apoptosis in rats exposed to acute ethanol intoxication.84 On the other hand, some death receptors and their ligands, especially Fas/FasL, have been found strongly expressed in hepatocytes of patients with alcoholic hepatitis compared with healthy controls or patients with alcoholic liver disease without hepatitis, which could increase the sensitivity of cytotoxic T lymphocyte mediated apoptosis.7,86 Hepatocyte apoptosis could also occur by autocrine and/or paracrine mechanisms, given the fact that both FasL and Fas are expressed on the same cell. Levels of circulating Fas and FasL were also found to be raised in patients with severe alcoholic hepatitis but the sources of these mediators and their biological importance remains to be investigated.87 The increase in FasL could be mediated by ROS,88 or may be the result of TNF-α induced activation of NFκB, which can upregulate the transcription of both Fas and FasL genes.89 Indeed, TNF-α serum levels are also increased during alcoholic hepatitis and play a crucial role in mediating hepatocyte damage.90 Chronic ethanol administration has also been shown to increase TNF-R expression in hepatocytes91 and therefore hepatocytes are likely to be more susceptible to apoptosis by TNF-α during alcohol exposure. Apart from a direct cytotoxic effect on the hepatocyte, the TNF-α/TNF-R1 system seems to be also required for Fas mediated cell death. Indeed, recent studies demonstrated that TNF-R1/TNF-R2 double knockout mice, which fail to undergo TNF-α mediated apoptosis, display increased resistance to Fas induced fulminant liver injury.92 Thus activation of the TNF-α/TNF-R1 complex may synergise with Fas mediated signalling to induce hepatocyte apoptosis, suggesting that both death receptors may contribute to ethanol mediated liver injury. Therapeutically, several studies have now been conduced in alcoholic hepatitis employing anti-TNF-α therapies. These biologicals, antibody therapeutics, appear promising although results are far from definitive.
Non-alcoholic steatohepatitis (NASH)
Non-alcoholic steatohepatitis (NASH) represents a subset of non-alcoholic fatty liver disease (NAFLD), characterised by the accumulation of fat in the liver (steatosis) along with inflammation in patients with no history of alcohol consumption or drug use/abuse. NASH can be associated with fibrosis and can progress to cirrhosis. The cellular mechanisms culminating in NASH remain poorly understood and therefore specific therapies for the treatment of this disease are still lacking. Recent studies demonstrated that hepatocyte apoptosis is increased in patients with non-alcoholic steatohepatitis, and correlates with disease severity and stage of fibrosis, suggesting an aetiopathogenic role for apoptosis in the progression of the disease.93,94 Death receptor expression, especially Fas and TNF-R1, is also significantly enhanced in patients with NASH, even more markedly than in alcoholic hepatitis patients.94,95 Thus NASH may sensitise hepatocytes to extracellular death ligands (that is, Fas, TNF-α), promoting apoptosis via the extrinsic pathway. Both Fas and TNF induced hepatocyte apoptosis proceeds via caspase-8 dependent cleavage of Bid, which translocates to mitochondria and induces mitochondrial dysfunction in cooperation with the other proapoptotic members Bax or Bak.39 Mitochondrial dysfunction results in release of cytochrome c, activation of effector caspases (caspase-3 and -7) and apoptosis. Consistently, liver samples from NASH patients have been found to show enhanced Fas expression, activation of caspase-3 and -7, and apoptosis, which positively correlated with biochemical and histopathological markers of liver injury.93–95 In patients with NASH, levels of circulating free fatty acids (FFA) are elevated and correlate with disease severity.96 Recent findings showed that treatment of liver cells in vitro with a mixture of long chain FFA resulted in Bax translocation to lysosomes and lysosomal destabilisation with release of lysosomal enzymes into the cytosol, which was, in part, dependent on a specific lysosomal enzyme, cathepsin B.97 Lysosomal permeabilisation was also confirmed in liver specimens from patients with NAFLD, with a positive correlation with disease severity. Lysosomal destabilisation resulted in NFκB dependent TNF-α expression, which promotes triglyceride accumulation and hepatic steatosis. Moreover, TNF-α can induce further lysosomal destabilisation and cathepsin B dependent apoptosis in a feed forward loop that exacerbates liver damage.98 The key role of cathepsin B in this process is demonstrated by data showing that, in a dietary murine model of NAFLD, either genetic or pharmacological inactivation of cathepsin B prevented the development of hepatic steatosis, liver injury, and insulin resistance.97 Consistently, genetic or pharmacological inhibition of cathepsin B has also been shown to reduce hepatocyte apoptosis and liver damage in steatotic liver after cold ischaemia/warm reperfusion injury.99 Thus therapeutic approaches aimed at inhibiting death receptor mediated apoptosis and/or cathepsin B may prove useful in reducing liver damage and preventing the development of cirrhosis in NASH.
Cholestatic liver disease
In cholestatic liver injury, elevated concentrations of bile acids accumulate in the tissue and within the hepatocytes as a consequence of reduced bile flow, and trigger liver injury. Although the mechanisms of liver damage associated with cholestasis are likely complex and multifactorial, bile acid mediated hepatotoxicity certainly plays a pivotal role in the pathogenesis of the disease. Hydrophobic bile acids have been shown to induce hepatocyte apoptosis in vitro8,100–102 and in vivo, in animal models of extrahepatic cholestasis.9,103 In particular, bile acids trigger hepatocyte apoptosis by activating the death receptor pathway in a ligand independent manner.8,101,104 Studies in vitro and in vivo have demonstrated that hepatocyte exposure to toxic bile acids results in ligand independent oligomerisation of Fas, recruitment of FADD, activation of caspase-8, and subsequent activation of effector proteases, including downstream caspases and cathepsin B. The importance of this pathway is underlined by reports that hepatocyte apoptosis is decreased in lpr mice, which express only minimal amounts of Fas, after bile duct ligation (an experimental model of extrahepatic cholestasis).9 The mechanism triggering Fas oligomerisation independent of FasL, however, is still unclear. Pathophysiological concentrations of conjugated bile acids have been shown to induce apoptosis only in cells expressing a bile acid transporter.105 Therefore, bile acids are likely to trigger receptor oligomerisation from inside the cell. In agreement with this hypothesis, elevated bile acid concentrations within the hepatocyte have been found to induce Fas translocation from its intracellular locations to the plasma membrane, where the increased surface density likely triggers its oligomerisation.100
Although Fas certainly plays a crucial role in bile acid mediated cytotoxicity, other death receptor pathways are also likely to be involved. The absence of functional Fas, indeed, seems to confer only transient protection, as suggested by the increase in apoptosis in lpr mice under conditions of persistent cholestasis.9 Studies on Fas deficient cells have demonstrated that at least one of these pathways involves transcriptional induction and oligomerisation of another death receptor, TRAIL-R2 (also called death receptor 5, DR5), suggesting other death receptor can functionally replace Fas in its absence.101 Because both Fas and TRAIL-R2 oligomerisation results in activation of caspase-8/-10 and Bid cleavage, targeted inhibition of caspases or Bid could be therapeutically useful in the treatment of cholestatic liver diseases. Consistently, experimental inhibition of Bid by injection of antisense oligonucleotides has been shown to reduce hepatocyte apoptosis and liver damage in bile duct ligated mice.103 Moreover, the pan-caspase inhibitor IDN-6556 has been found to be effective in attenuating liver injury and fibrosis in bile duct ligated mice.106 The drug is currently in clinical trial for treatment of various liver diseases.107
Hepatic fibrosis and cirrhosis
Fibrogenesis is a relatively late event in chronic liver injury, and occurs as a consequence of activation of hepatic stellate cells (HSC) and excessive deposition of extracellular matrix, under conditions of persistent inflammation. The first phase of liver injury, however, independent of the aetiology, is almost always characterised by increased hepatocyte apoptosis. Because for many years apoptosis has been considered a mechanism of cell death not associated with an inflammatory response, the two phases in the development of the disease have never been connected. It is only recently that a link between these two pathological events has been established.108 Recent studies have demonstrated that hepatic fibrosis was significantly reduced in a model of experimental extrahepatic cholestasis when Fas mediated apoptosis was impaired, or when caspases or cathepsin B, a lysosomal enzyme involved in bile acid induced apoptosis, were inhibited.10,106,109 Conversely, persistent hepatocyte apoptosis due to hepatocyte specific disruption of Bcl-XL has been shown to lead to liver fibrosis with advanced age.110 This latter model is highly illustrative because it directly demonstrates that hepatocyte apoptosis is profibrogenic.
Apoptosis is indeed a proinflammatory process when occurs in pathological conditions. In the presence of massive hepatocyte apoptosis, the ability of phagocytic cells to effectively and rapidly remove dead cells in tissue is overwhelmed, with accumulation and subsequent autolysis of the apoptotic bodies, and release of their proinflammatory contents. Moreover, engulfment of apoptotic bodies by Kupffer cells, the major phagocytes in the liver, has been demonstrated to enhance expression of death ligands, especially Fas ligand and the proinflammatory cytokine TNF-α, thereby accelerating hepatocyte apoptosis and eliciting hepatic inflammation.111 Phagocytosis of apoptotic bodies also promotes generation of transforming growth factor β (TGF-β), a cytokine with potent profibrogenic and proapoptotic activity in the liver.110,112,113 Although their capacity to engulf apoptotic bodies is lower than Kupffer cells, HSC are anatomically better positioned to engulf apoptotic bodies from dying hepatocytes than Kupffer cells, and have been shown to clear apoptotic bodies in vitro.114 More importantly, this process is associated with activation of quiescent HSC, as demonstrated by induction of the classic marker α-smooth muscle actin, and increase production of TGF-β1 and collagen Ia, both markers of fibrogenic activity.114 More data however are required to ascertain the occurrence of the process in vivo. Thus it appears that engulfment of apoptotic bodies by both Kupffer cells and HSC promotes their expression of profibrogenic proteins and death ligands. Persistent activation of these cells results in exacerbation of hepatocyte apoptosis, hepatic inflammation, sustained HSC activation, and progression to liver cirrhosis (fig 4 ▶). Therefore, purposeful induction of apoptosis of activated HSC may be a useful antifibrotic therapy. As activated HSC have been found to express preferentially TRAIL-R2 and show increased sensitivity to TRAIL mediated apoptosis,115 whereas TRAIL-R2 is not expressed on hepatocytes, induction of apoptosis via treatment with a TRAIL-R2 agonist antibody might represent an ideal therapeutic strategy to selectively kill HSC. Inhibition of apoptotic body engulfment by HSC, or modulation of signalling events occurring in HSC as a result of phagocytosis of apoptotic bodies, also appear to be potentially valid therapeutic strategies to inhibit liver fibrogenesis.
Figure 4.
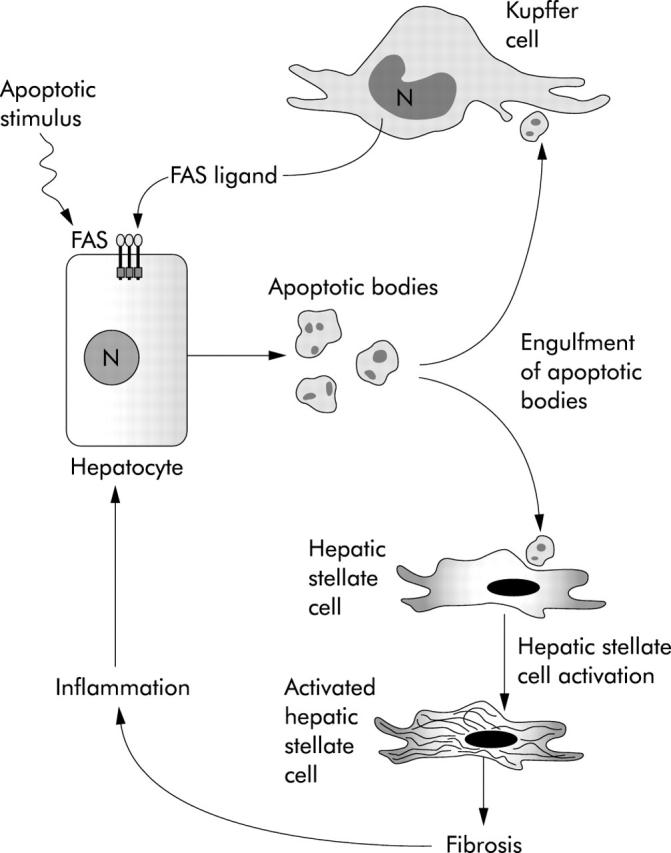
The link between apoptosis and fibrosis in the liver. Schematic representation of the molecular mechanisms involved in the proposed model linking hepatocyte apoptosis and liver fibrosis. Different injuries on the liver result in hepatocyte apoptosis. The generated apoptotic bodies are cleared by phagocytosis by Kupffer cells and hepatic stellate cells, which enhances their expression of profibrogenic genes and death ligands, such as Fas ligand. Persistent activation of these cells promotes further hepatocyte apoptosis, progression of fibrosis, hepatic inflammation, and liver damage.
SUMMARY AND CONCLUDING REMARKS
Apoptosis represents the physiological way to eliminate excessive cells during embryogenesis and tissue remodelling. Under these conditions, apoptosis occurs in a controlled environment where dying cells are promptly removed by phagocytosis and replaced by new cells generated by mitosis. Apoptosis, however, is also an essential feature of a wide variety of acute and chronic diseases, including liver diseases. Imbalance between cell proliferation and death always leads to loss of tissue homeostasis and onset of various diseases. Excessive apoptosis has, indeed, been identified in acute and chronic viral hepatitis, alcoholic and non-alcoholic hepatitis, cholestatic liver disease, Wilson’s disease, and graft versus host disease (GVHD). Sustained apoptosis has also been linked with the development of hepatic fibrosis. In contrast, insufficient apoptosis has been associated with development and progression of tumours of the liver and the biliary tree. Thus identification of target molecules involved in apoptosis may offer new options for pharmacological and/or gene mediated therapies for patients with liver diseases (fig 5 ▶).
Figure 5.

Therapeutic modulation of apoptosis in liver diseases. Imbalance between cell proliferation and cell death in the liver contributes to the pathogenesis of several liver diseases. Excessive apoptosis is associated with acute diseases, such as acute and fulminant hepatitis, as well as with chronic diseases, such as chronic hepatitis, alcoholic liver disease, cholestatic liver disease, and non-alcoholic steatohepatitis (NASH). Sustained apoptosis also causes persistent inflammation and promotes fibrogenesis. In contrast, deficient apoptosis contributes to the development of liver and biliary cancer. Therapeutic strategies currently in use or potentially useful to modulate apoptosis in liver diseases are depicted. TRAIL, tumour necrosis factor related apoptosis inducing ligand (TRAIL); HSC, hepatic stellate cells; siRNA, small interfering RNA.
Acknowledgments
This work was supported by NIH grant (DK 41876 to GJG), the Palumbo Foundation, and the Mayo Foundation.
Conflict of interest: None declared.
REFERENCES
- 1.Oda T, Tsuda H, Scarpa A, et al. p53 gene mutation spectrum in hepatocellular carcinoma. Cancer Res 1992;52:6358–64. [PubMed] [Google Scholar]
- 2.Que FG, Phan VA, Phan VH, et al. Cholangiocarcinomas express Fas ligand and disable the Fas receptor. Hepatology 1999;30:1398–404. [DOI] [PubMed] [Google Scholar]
- 3.Nzeako UC, Guicciardi ME, Yoon JH, et al. COX-2 inhibits Fas-mediated apoptosis in cholangiocarcinoma cells. Hepatology 2002;35:552–9. [DOI] [PubMed] [Google Scholar]
- 4.Pikarsky E, Porat RM, Stein I, et al. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004;431:461–6. [DOI] [PubMed] [Google Scholar]
- 5.Ogasawara J, Watanabe-Fukunaga R, Adachi M, et al. Lethal effect of the anti-Fas antibody in mice. Nature 1993;364:806–9. [DOI] [PubMed] [Google Scholar]
- 6.Kohli V, Selzner M, Madden JF, et al. Endothelial cell and hepatocyte deaths occur by apoptosis after ischemia-reperfusion injury in the rat liver. Transplantation 1999;67:1099–105. [DOI] [PubMed] [Google Scholar]
- 7.Natori S, Rust C, Stadheim LM, et al. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J Hepatol 2001;34:248–53. [DOI] [PubMed] [Google Scholar]
- 8.Faubion WA, Guicciardi ME, Miyoshi H, et al. Toxic bile salts induce rodent hepatocyte apoptosis via direct activation of Fas. J Clin Invest 1999;103:137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyoshi H, Rust C, Roberts PJ, et al. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999;117:669–77. [DOI] [PubMed] [Google Scholar]
- 10.Canbay A, Higuchi H, Bronk SF, et al. Fas enhances fibrogenesis in the bile duct ligated mouse: a link between apoptosis and fibrosis. Gastroenterology 2002;123:1323–30. [DOI] [PubMed] [Google Scholar]
- 11.Patel T, Gores GJ. Apoptosis and hepatobiliary disease. Hepatology 1995;21:1725–41. [DOI] [PubMed] [Google Scholar]
- 12.Patel T, Roberts LR, Jones BA, et al. Dysregulation of apoptosis as a mechanism of liver disease: an overview. Semin Liver Dis 1998;18:105–14. [DOI] [PubMed] [Google Scholar]
- 13.Rust C, Gores GJ. Apoptosis and liver disease. Am J Med 2000;108:567–74. [DOI] [PubMed] [Google Scholar]
- 14.Yoon JH, Gores GJ. Death receptor-mediated apoptosis and the liver. J Hepatol 2002;37:400–10. [DOI] [PubMed] [Google Scholar]
- 15.Jacobson MD, Weil M, Raff MC. Programmed cell death in animal development. Cell 1997;88:347–54. [DOI] [PubMed] [Google Scholar]
- 16.Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int Rev Cytol 1980;68:251–306. [DOI] [PubMed] [Google Scholar]
- 17.Takehara T, Liu X, Fujimoto J, et al. Expression and role of Bcl-xL in human hepatocellular carcinomas. Hepatology 2001;34:55–61. [DOI] [PubMed] [Google Scholar]
- 18.Bantel H, Lugering A, Heidemann J, et al. Detection of apoptotic caspase activation in sera from patients with chronic HCV infection is associated with fibrotic liver injury. Hepatology 2004;40:1078–87. [DOI] [PubMed] [Google Scholar]
- 19.Lauber K, Bohn E, Krober SM, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 2003;113:717–30. [DOI] [PubMed] [Google Scholar]
- 20.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science 1998;281:1305–8. [DOI] [PubMed] [Google Scholar]
- 21.Green DR, Reed JC. Mitochondria and apoptosis. Science 1998;281:1309–12. [DOI] [PubMed] [Google Scholar]
- 22.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science 2004;305:626–9. [DOI] [PubMed] [Google Scholar]
- 23.Nicholson DW, Thornberry NA. Caspases: killer proteases. Trends Biochem Sci 1997;22:299–306. [DOI] [PubMed] [Google Scholar]
- 24.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell 2004;117:561–74. [DOI] [PubMed] [Google Scholar]
- 25.Cory S, Adams JM. The Bcl-2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer 2002;2:647–56. [DOI] [PubMed] [Google Scholar]
- 26.Cory S, Huang DC, Adams JM. The Bcl-2 family: roles in cell survival and oncogenesis. Oncogene 2003;22:8590–607. [DOI] [PubMed] [Google Scholar]
- 27.Antonsson B, Conti F, Ciavatta A, et al. Inhibition of Bax channel-forming activity by Bcl-2. Science 1997;277:370–2. [DOI] [PubMed] [Google Scholar]
- 28.Hsu Y-T, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem 1998;272:10777–83. [DOI] [PubMed] [Google Scholar]
- 29.Antonsson B, Montessuit S, Sanchez B, et al. Bax is present as a high molecular weight oligomer/complex in the mitochondrial membrane of apoptotic cells. J Biol Chem 2001;276:11615–23. [DOI] [PubMed] [Google Scholar]
- 30.Nechushtan A, Smith CL, Lamensdorf I, et al. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J Cell Biol 2001;153:1265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei MC, Lindsten T, Mootha VK, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev 2000;14:2060–71. [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Yin XM, Chao DT, et al. BID: a novel BH3 domain-only death agonist. Genes Dev 1996;10:2859–69. [DOI] [PubMed] [Google Scholar]
- 33.Cheng EH, Wei MC, Weiler S, et al. BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 2001;8:705–11. [DOI] [PubMed] [Google Scholar]
- 34.Wei MC, Zong WX, Cheng EH, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001;292:727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo X, Budihardjo I, Zou H, et al. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998;94:481–90. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Zhu H, Xu CJ, et al. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998;94:491–501. [DOI] [PubMed] [Google Scholar]
- 37.Faubion WA, Gores GJ. Death receptors in liver biology and pathobiology. Hepatology 1999;29:1–4. [DOI] [PubMed] [Google Scholar]
- 38.Krammer PH. CD95’s deadly mission in the immune system. Nature 2000;407:789–95. [DOI] [PubMed] [Google Scholar]
- 39.Scaffidi C, Fulda S, Srinivasan A, et al. Two CD95 (APO-1/Fas) signaling pathways. Embo J 1998;17:1675–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yin XM, Wang K, Gross A, et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999;400:886–91. [DOI] [PubMed] [Google Scholar]
- 41.Rocken C, Carl-McGrath S. Pathology and pathogenesis of hepatocellular carcinoma. Dig Dis 2001;19:269–78. [DOI] [PubMed] [Google Scholar]
- 42.Hollstein M, Sidransky D, Vogelstein B, et al. p53 mutations in human cancers. Science 1991;253:49–53. [DOI] [PubMed] [Google Scholar]
- 43.Harris CC, Hollstein M. Clinical implications of the p53 tumor suppressor gene. N Engl J Med 1993;329:1318–27. [DOI] [PubMed] [Google Scholar]
- 44.Liu X, Yue P, Khuri FR, et al. p53 upregulates death receptor 4 expression through an intronic p53 binding site. Cancer Res 2004;64:5078–83. [DOI] [PubMed] [Google Scholar]
- 45.Lin P, Bush JA, Cheung KJJ, et al. Tissue-specific regulation of Fas/APO-1/CD95 expression by p53. Int J Oncol 2002;21:261–4. [PubMed] [Google Scholar]
- 46.Muller M, Wilder S, Bannasch D, et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 1998;188:2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell 2003;11:577–90. [DOI] [PubMed] [Google Scholar]
- 48.Anderson SC, Johnson DE, Harris MP, et al. p53 gene therapy in a rat model of hepatocellular carcinoma: intra-arterial delivery of a recombinant adenovirus. Clin Cancer Res 1998;4:1649–59. [PubMed] [Google Scholar]
- 49.Horowitz J. Adenovirus-mediated p53 gene therapy: overview of preclinical studies and potential clinical applications. Curr Opin Mol Ther 1999;1:500–9. [PubMed] [Google Scholar]
- 50.Makower D, Rozenblit A, Kaufman H, et al. Phase II clinical trial of intralesional administration of the oncolytic adenovirus ONYX-015 in patients with hepatobiliary tumors with correlative p53 studies. Clin Cancer Res 2003;9:693–702. [PubMed] [Google Scholar]
- 51.Ruiz J, Mazzolini G, Sangro B, et al. Gene therapy of hepatocellular carcinoma. Dig Dis 2001;19:324–32. [DOI] [PubMed] [Google Scholar]
- 52.Leithauser F, Dhein J, Mechtersheimer G, et al. Constitutive and induced expression of APO-1, a new member of the NGF/TNF receptor superfamily, in normal and neoplastic cells. Lab Invest 1993;69:415–29. [PubMed] [Google Scholar]
- 53.Higaki K, Yano H, Kojiro M. Fas antigen expression and its relationship with apoptosis in human hepatocellular carcinoma and noncancerous tissues. Am J Pathol 1996;149:429–37. [PMC free article] [PubMed] [Google Scholar]
- 54.Hahne M, Rimoldi D, Schroter M, et al. Melanoma cell expression of Fas(Apo-1/CD95) ligand: implications for tumor immune escape. Science 1996;274:1363–6. [DOI] [PubMed] [Google Scholar]
- 55.Strand S, Hofmann WJ, Hug H, et al. Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells—a mechanism of immune evasion? Nature Med 1996;2:1361–6. [DOI] [PubMed] [Google Scholar]
- 56.Griffith TS, Brunner T, Fletcher SM, et al. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995;17:1189–92. [DOI] [PubMed] [Google Scholar]
- 57.Ito Y, Takeda T, Umeshita K, et al. Fas antigen expression in hepatocellular carcinoma tissues. Oncol Rep 1998;5:41–4. [PubMed] [Google Scholar]
- 58.Nagao M, Nakajima Y, Hisanaga M, et al. The alteration of Fas receptor and ligand system in hepatocellular carcinomas: how do hepatoma cells escape from the host immune surveillance in vivo? Hepatology 1999;30:413–21. [DOI] [PubMed] [Google Scholar]
- 59.Fukuzawa Y, Takahashi K, Furuta K, et al. Expression of Fas/Fas ligand and its involvement in infiltrating lymphocytes in hepatocellular carcinoma (HCC). J Gastroenterol 2001;36:681–8. [DOI] [PubMed] [Google Scholar]
- 60.Muller M, Strand S, Hug H, et al. Drug-induced apoptosis in hepatoma cells is mediated by the CD95 (APO-1/Fas) receptor/ligand system and involves activation of wild-type p53. J Clin Invest 1997;99:403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiramatsu N, Hayashi N, Katayama K, et al. Immunohistochemical detection of Fas antigen in liver tissue of patients with chronic hepatitis C. Hepatology 1994;19:1354–9. [PubMed] [Google Scholar]
- 62.Mita E, Hayashi N, Iio S, et al. Role of Fas ligand in apoptosis induced by hepatitis C virus infection. Biochem Biophys Res Com 1994;204:468–74. [DOI] [PubMed] [Google Scholar]
- 63.Yoneyama K, Goto T, Miura K, et al. The expression of Fas and Fas ligand, and the effects of interferon in chronic liver diseases with hepatitis C virus. Hepatol Res 2002;24:327–37. [DOI] [PubMed] [Google Scholar]
- 64.Mochizuki K, Hayashi N, Hiramatsu N, et al. Fas antigen expression in liver tissue of patients with chronic hepatitis B. J Hepatol 1996;24:1–7. [DOI] [PubMed] [Google Scholar]
- 65.Luo KX, Zhu YF, Zhang LX, et al. In situ investigation of Fas/FasL expression in chronic hepatitis B virus infection and its related liver diseases. J Viral Hep 1997;4:303–7. [DOI] [PubMed] [Google Scholar]
- 66.Galle PR, Hofmann WJ, Walczak H, et al. Involvement of the APO-1/Fas (CD95) receptor and ligand in liver damage. J Exp Med 1995;182:1223–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lowin B, Hahne M, Mattmann C, et al. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathyways. Nature 1994;370:650–2. [DOI] [PubMed] [Google Scholar]
- 68.Kagi D, Vignaux F, Ledermann B, et al. Fas and perforin pathways as major mechanisms of T-cell-mediated cytotoxicity. Science 1994;265:528–30. [DOI] [PubMed] [Google Scholar]
- 69.Bantel H, Schulze-Osthoff K. Apoptosis in hepatitis C virus infection. Cell Death Differ 2003;10 (suppl 1) :S48–58. [DOI] [PubMed] [Google Scholar]
- 70.Kafrouni MI, Brown GR, Thiele DL. Virally infected hepatocytes are resistant to perforin-dependent CTL effector mechanisms. J Immunol 2001;167:1566–74. [DOI] [PubMed] [Google Scholar]
- 71.Terradillos O, De La Coste A, Pollicino T, et al. The hepatitis B virus X protein abrogates Bcl-2-mediated protection against Fas apoptosis in the liver. Oncogene 2002;21:377–86. [DOI] [PubMed] [Google Scholar]
- 72.Pan J, Duan LX, Sun BS, et al. Hepatitis B virus X protein protects against anti-Fas-mediated apoptosis in human liver cells by inducing NF-κB. J Gen Virol 2001;82:171–82. [DOI] [PubMed] [Google Scholar]
- 73.Diao J, Khine AA, Sarangi F, et al. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J Biol Chem 2001;276:8328–40. [DOI] [PubMed] [Google Scholar]
- 74.Ruggieri A, Harada T, Matsuura Y, et al. Sensitization to Fas-mediated apoptosis by hepatitis C virus core protein. Virology 1997;229:68–76. [DOI] [PubMed] [Google Scholar]
- 75.Machida K, Tsukiyama-Kohara K, Seike E, et al. Inhibition of cytochrome c release in Fas-mediated signaling pathway in transgenic mice induced to express hepatitis C viral proteins. J Biol Chem 2001;276:12140–6. [DOI] [PubMed] [Google Scholar]
- 76.Zender L, Hutker S, Liedtke C, et al. Caspase-8 small interfering RNA prevents acute liver failure in mice. Proc Natl Acad Sci U S A 2003;100:7797–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Song E, Lee SK, Wang J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nature Med 2003;9:347–51. [DOI] [PubMed] [Google Scholar]
- 78.Fiorucci S, Mencarelli A, Palazzetti B, et al. An NO derivative of ursodeoxycholic acid protects against Fas-mediated liver injury by inhibiting caspase activity. Proc Natl Acad Sci U S A 2001;98:2652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Goldin RD, Hunt NC, Clark J, et al. Apoptotic bodies in a murine model of alcoholic liver disease. J Pathol 1993;171:73–6. [DOI] [PubMed] [Google Scholar]
- 80.Benedetti A, Brunelli E, Risicate R, et al. Subcellular changes and apoptosis induced by ethanol in rat liver. J Hepatol 1988;6:137–43. [DOI] [PubMed] [Google Scholar]
- 81.Ziol M, Tepper M, Lohez M, et al. Clinical and biological relevance of hepatocyte apoptosis in alcoholic hepatitis. J Hepatol 2001;34:254–60. [DOI] [PubMed] [Google Scholar]
- 82.Jaeschke H. Neutrophil-mediated tissue injury in alcoholic hepatitis. Alcohol 2002;27:23–7. [DOI] [PubMed] [Google Scholar]
- 83.Jaeschke H. Inflammation in response to hepatocellular apoptosis. Hepatology 2002;35:964–6. [DOI] [PubMed] [Google Scholar]
- 84.Kurose I, Higuchi H, Miura S, et al. Oxidative stress-mediated apoptosis of hepatocytes exposed to acute ethanol intoxication. Hepatology 1997;25:368–78. [DOI] [PubMed] [Google Scholar]
- 85.French SW, Wong K, Jui L, et al. Effect of ethanol on cytochrome P450 2E1 lipid peroxidation, and serum protein adduct formation in relation to liver pathology pathogenesis. Exp Mol Pathol 1993;58:61–75. [DOI] [PubMed] [Google Scholar]
- 86.Tagami A, Ohnishi H, Moriwaki H, et al. Fas-mediated apoptosis in acute alcoholic hepatitis. Hepatogastroenteroloy 2003;50:443–8. [PubMed] [Google Scholar]
- 87.Taieb J, Mathurin P, Poynard T, et al. Raised plasma soluble Fas and Fas-ligand in alcoholic liver disease. Lancet 1998;351:1930–1. [DOI] [PubMed] [Google Scholar]
- 88.Hug H, Strand S, Grambihler A, et al. Reactive oxygen intermediates are involved in the induction of CD95 ligand mRNA expression by cytostatic drugs in hepatoma cells. J Biol Chem 1997;272:28191–3. [DOI] [PubMed] [Google Scholar]
- 89.Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50–p65 recruitment. Mol Cell Biol 1999;19:2098–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McClain C, Hill D, Schmidt J, et al. Cytokines and alcoholic liver disease. Semin Liver Dis 1993;13:170–82. [DOI] [PubMed] [Google Scholar]
- 91.Deaciuc IV, D’Souza NB, Spitzer JJ. Tumor necrosis factor-alpha cell-surface receptors of liver parenchymal and nonparenchymal cells during acute and chronic alcohol administration to rats. Alcohol Clin Exp Res 1995;19:332–8. [DOI] [PubMed] [Google Scholar]
- 92.Costelli P, Aoki P, Zingaro B, et al. Mice lacking TNFα receptors 1 and 2 are resistant to death and fulminant liver injury induced by agonistic anti-Fas antibody. Cell Death Differ 2003;10:997–1004. [DOI] [PubMed] [Google Scholar]
- 93.Ribeiro PS, Cortez-Pinto H, Sola S, et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. Am J Gastroenterol 2004;99:1708–17. [DOI] [PubMed] [Google Scholar]
- 94.Feldstein AE, Canbay A, Angulo P, et al. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003;125:437–43. [DOI] [PubMed] [Google Scholar]
- 95.Feldstein AE, Canbay A, Guicciardi ME, et al. Diet associated hepatic steatosis sensitizes to Fas mediated liver injury in mice. J Hepatol 2003;39:978–83. [DOI] [PubMed] [Google Scholar]
- 96.Nehra V, Angulo P, Buchman A, et al. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Dig Dis Sci 2001;46:2347–52. [DOI] [PubMed] [Google Scholar]
- 97.Feldstein AE, Werneburg NW, Canbay A, et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004;40:185–94. [DOI] [PubMed] [Google Scholar]
- 98.Guicciardi ME, Deussing J, Miyoshi H, et al. Cathepsin B contributes to TNF-alpha-mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J Clin Invest 2000;106:1127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Baskin-Bey ES, Canbay A, Bronk SF, et al. Cathepsin B inactivation attenuates hepatocyte apoptosis and liver damage in steatotic livers after cold ischemia/warm reperfusion injury. Am J Physiol Gastrointest Liver Physiol 2005;288:G396–402. [DOI] [PubMed] [Google Scholar]
- 100.Sodeman T, Bronk SF, Roberts PJ, et al. Bile salts mediate hepatocyte apoptosis by increasing cell surface trafficking of Fas. Am J Physiol Gastrointest Liver Physiol 2000;278:G992–9. [DOI] [PubMed] [Google Scholar]
- 101.Higuchi H, Bronk SF, Takikawa Y, et al. The bile acid glycochenodeoxycholate induces trail-receptor 2/DR5 expression and apoptosis. J Biol Chem 2001;276:38610–18. [DOI] [PubMed] [Google Scholar]
- 102.Guicciardi ME, Gores GJ. Bile acid-mediated hepatocyte apoptosis and cholestatic liver disease. Dig Liver Dis 2002;34:387–92. [DOI] [PubMed] [Google Scholar]
- 103.Higuchi H, Miyoshi H, Bronk SF, et al. Bid antisense attenuates bile acid-induced apoptosis and cholestatic liver injury. J Pharmacol Exp Ther 2001;299:866–73. [PubMed] [Google Scholar]
- 104.Higuchi H, Bronk SF, Taniai M, et al. Cholestasis increases tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-R2/DR5 expression and sensitizes the liver to TRAIL-mediated cytotoxicity. J Pharmacol Exp Ther 2002;303:461–7. [DOI] [PubMed] [Google Scholar]
- 105.Torchia EC, Stolz A, Agellon LB. Differential modulation of cellular death and survival pathways by conjugated bile acids. BMC Biochem 2001;2:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Canbay A, Feldstein A, Baskin-Bey E, et al. The caspase inhibitor IDN-6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther 2004;308:1191–6. [DOI] [PubMed] [Google Scholar]
- 107.Valentino KL, Gutierrez M, Sanchez R, et al. First clinical trial of a novel caspase inhibitor: anti-apoptotic caspase inhibitor, IDN-6556, improves liver enzymes. Int J Clin Pharmacol Ther 2003;41:441–9. [DOI] [PubMed] [Google Scholar]
- 108.Canbay A, Friedman S, Gores GJ. Apoptosis: the nexus of liver injury and fibrosis. Hepatology 2004;39:273–8. [DOI] [PubMed] [Google Scholar]
- 109.Canbay A, Guicciardi ME, Higuchi H, et al. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Clin Invest 2003;112:152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Takehara T, Tatsumi T, Suzuki T, et al. Hepatocyte-specific disruption of Bcl-xL leads to continuous hepatocyte apoptosis and liver fibrotic responses. Gastroenterology 2004;127:1189–97. [DOI] [PubMed] [Google Scholar]
- 111.Canbay A, Feldstein AE, Higuchi H, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003;38:1188–98. [DOI] [PubMed] [Google Scholar]
- 112.Friedman SL. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 2000;275:2247–50. [DOI] [PubMed] [Google Scholar]
- 113.Oberhammer FA, Pavelka M, Sharma S, et al. Induction of apoptosis in cultured hepatocytes and in regressing liver by transforming growth factor beta-1. Proc Natl Acad Sci U S A 1992;89:5408–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Canbay A, Taimr P, Torok N, et al. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest 2003;83:655–63. [DOI] [PubMed] [Google Scholar]
- 115.Taimr P, Higuchi H, Kocova E, et al. Activated stellate cells express the TRAIL receptor-2/death receptor-5 and undergo TRAIL-mediated apoptosis. Hepatology 2003;37:87–95. [DOI] [PubMed] [Google Scholar]
- 116.Ito Y, Monden M, Takeda T, et al. The status of Fas and Fas ligand expression can predict recurrence of hepatocellular carcinoma. Br J Cancer 2000;82:1211–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lee SH, Shin MS, Lee HS, et al. Expression of Fas and Fas-related molecules in human hepatocellular carcinoma. Hum Pathol 2001;32:250–6. [DOI] [PubMed] [Google Scholar]
- 118.Okano H, Shiraki K, Inoue H, et al. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest 2003;83:1033–43. [DOI] [PubMed] [Google Scholar]
- 119.Fiore G, Piazzolla G, Galetta V, et al. Liver tissue expression of CD80 and CD95 antigens in chronic hepatitis C: relationship with biological and histological disease activities. Microbios 1999;97:29–38. [PubMed] [Google Scholar]
- 120.Tagashira M, Yamamoto K, Fujio K, et al. Expression of perforin and Fas ligand mRNA in the liver of viral hepatitis. J Clin Immunol 2000;20:347–53. [DOI] [PubMed] [Google Scholar]
- 121.Ehrmann J Jr, Galuszkova D, Ehrmann J, et al. Apoptosis-related proteins, BCL-2, BAX, FAS, FAS-L and PCNA in liver biopsies of patients with chronic hepatitis B virus infection. Pathol Oncol Res 2000;6:130–5. [DOI] [PubMed] [Google Scholar]
- 122.Pianko S, Patella S, Ostapowicz G, et al. Fas-mediated hepatocyte apoptosis is increased by hepatitis C virus infection and alcohol consumption, and may be associated with hepatic fibrosis: mechanisms of liver cell injury in chronic hepatitis C virus infection. J Viral Hepat 2001;8:406–13. [DOI] [PubMed] [Google Scholar]
- 123.Bantel H, Lugering A, Poremba C, et al. Caspase activation correlates with the degree of inflammatory liver injury in chronic hepatitis C virus infection. Hepatology 2001;34:758–67. [DOI] [PubMed] [Google Scholar]
- 124.Ibuki N, Yamamoto K, Yabushita K, et al. In situ expression of Granzime B and Fas-ligand in the liver of viral hepatitis. Liver 2002;22:198–204. [DOI] [PubMed] [Google Scholar]
- 125.Tang TJ, Kwekkeboom J, Laman JD, et al. The role of intrahepatic immune effector cells in inflammatory liver injury and viral control during chronic hepatitis B infection. J Viral Hepatol 2003;10:159–67. [DOI] [PubMed] [Google Scholar]
- 126.Rivero M, Crespo J, Fabrega E, et al. Apoptosis mediated by the Fas system in the fulminant hepatitis by hepatitis B virus. J Viral Hepatol 2002;9:107–13. [DOI] [PubMed] [Google Scholar]
- 127.Ryo K, Kamogawa Y, Ikeda I, et al. Significance of Fas antigen-mediated apoptosis in human fulminant hepatic failure. Am J Gastroenterol 2000;95:2047–55. [DOI] [PubMed] [Google Scholar]
- 128.Fox CK, Furtwaengler A, Nepomuceno RR, et al. Apoptotic pathways in primary biliary cirrhosis and autoimmune hepatitis. Liver 2001;21:272–9. [DOI] [PubMed] [Google Scholar]
- 129.Strand S, Hofmann WJ, Grambihler A, et al. Hepatic failure and liver cell damage in acute Wilson’s disease involve CD95 (APO-1/Fas) mediated apoptosis. Nat Med 1998;4:588–93. [DOI] [PubMed] [Google Scholar]
- 130.Tannapfel A, Kohlhaw K, Ebelt J, et al. Apoptosis and the expression of Fas and Fas ligand (FasL) antigen in rejection and reinfection in liver allograft specimens. Transplantation 1999;67:1079–83. [DOI] [PubMed] [Google Scholar]