

Abstract
Background: Transferrin receptor 2 (TfR2) is a key molecule involved in the regulation of iron homeostasis. Mutations in humans cause type 3 haemochromatosis and a targeted mutation in mice leads to iron overload with a similar phenotype. We have previously described the generation of a complete TfR2-knockout (KO) mouse.
Aims: The aims of this study were to determine the phenotype and analyse expression of iron related molecules in the liver, duodenum, and spleen of homozygous TfR2-KO, heterozygous, and wild-type mice.
Methods: Serum and tissue iron levels were determined in 10 week old male mice. Expression of iron related mRNA transcripts were analysed in the liver, duodenum, and spleen using real time polymerase chain reaction. Expression of iron related proteins in the liver were analysed by immunoblotting and immunohistochemistry.
Results: Homozygous TfR2-KO mice had no TfR2 protein expression and developed significant iron overload typical of TfR2 associated haemochromatosis. In the liver of TfR2-KO mice there was no upregulation of hepcidin mRNA or prohepcidin protein in response to iron loading.
Conclusions: Our results suggest that TfR2 is required for iron regulated expression of hepcidin and is involved in a pathway related to Hfe and hemojuvelin.
Keywords: transferrin receptor 2, hepcidin, haemochromatosis, iron metabolism, knockout mouse
Transferrin receptor 2 (TfR2) is a homologue of transferrin receptor 1 (TfR1), the primary molecule responsible for the uptake of transferrin bound iron into cells.1 Unlike the ubiquitously expressed TfR1, TfR2 is thought to be expressed almost exclusively in the liver and erythroid cells.1TfR2 mRNA contains no iron responsive elements and does not appear to be regulated by iron.2 TfR2 can bind transferrin but at a much reduced affinity compared with TfR1.3,4 The exact role of TfR2 in iron metabolism is unknown although it is evident that it plays an important function in maintaining iron homeostasis as mutations in the TfR2 gene lead to haemochromatosis in humans.5,6 Mice with a targeted mutation of TfR2 (Y245X, orthologous to human Y250X) develop iron overload with a similar phenotype to patients with TfR2 associated haemochromatosis.7
Hepcidin, originally described as an antimicrobial peptide, has been shown to be a key regulator of iron homeostasis.8,9,10 Hepcidin is produced in the liver and is secreted into the circulation in response to inflammation or increased iron stores.10,11 Hepcidin is a negative regulator of iron absorption and iron release from cells. Mice have two hepcidin genes located adjacent to each other on chromosome 7.10 This differs from humans and rats which only have one gene. Mutations of hepcidin in humans and knockout in mice lead to a severe form of haemochromatosis.12,13 Regulation of hepcidin appears to be impaired in hereditary haemochromatosis. In patients with HFE associated haemochromatosis and in Hfe knockout mice, there is loss of regulation of hepcidin mRNA expression, causing hepcidin levels to remain low despite increased iron stores.14,15 Therefore, it appears that HFE is involved in the iron induced pathway of hepcidin expression. One study has suggested that HFE is also involved in the inflammation induced pathway,16 but this is controversial, and other groups have shown that the inflammation induced pathway is independent of HFE17,18 and that this pathway is also independent of TfR2.18 The recently identified molecule mutated in juvenile haemochromatosis, hemojuvelin (Hjv), is also thought to be a key player in the regulation of hepcidin expression.19 However, the location of Hjv within the hepcidin regulatory pathway is not yet known.
Previous studies have shown that targeted mutations and knockout of genes in mice do not always produce exactly the same phenotype, as some mutant proteins can retain partial function. For example, mice with a full knockout of the Hfe gene develop more severe iron overload than mice with the targeted Hfe-C282Y mutation (the primary cause of HFE associated haemochromatosis in humans), suggesting that the C282Y mutation may not completely abrogate the function of HFE.20 Therefore, to complement studies on mice with the targeted TfR2-Y245X mutation, we have generated and studied mice with a full knockout of the TfR2 gene. We used a Cre recombinase:loxP strategy to delete exons 2–6 of the mouse TfR2 gene.21 These mice have complete absence of TfR2 protein expression, as determined by immunoblotting, with TfR2 antibodies raised against both the amino terminal cytoplasmic domain and the carboxyl terminal extracellular domain. We have used these mice to investigate the molecular basis of iron overload and study expression of iron related molecules in the liver, duodenum, and spleen.
METHODS
Mice
Mice with a deletion of exons 2–6 of the TfR2 gene were generated using the Cre recombinase:loxP system, as previously described.21 Heterozygous TfR2-knockout (KO) mice on a mixed 129/C57BL6/J strain background were intercrossed to produce homozygous, heterozygous, and wild-type mice. Ten or 11 male mice from each group were sacrificed at 10 weeks of age and tissues and blood were taken for further analysis. All experiments were performed using littermates to avoid any strain background effects. All mice were maintained on standard laboratory chow (Norco Stockfeeds, South Lismore, Australia). Animals received humane care and study protocols were approved by the Queensland Institute of Medical Research Animal Ethics Committee.
Antibodies
Anti-TfR1 antibodies were purchased from Zymed Laboratories (San Francisco, California, USA), antiferritin antibodies from the Binding Site (UK), and antiactin antibodies from Sigma-Aldrich (Castle Hill, Australia). Antibodies against mTfR2 were generated in rabbits using a recombinant protein comprising the amino terminal 81 amino acids of the mouse TfR2 cytoplasmic domain. Antiprohepcidin antibodies were generated using recombinant GST-mouse prohepcidin corresponding to amino acids 19–83.22
Measurement of iron indices
Transferrin saturation was measured using an iron and iron binding capacity kit (Sigma-Aldrich). Liver iron concentration was measured using the method of Torrance and Bothwell.23 The Perls’ Prussian blue staining method was used for detecting iron in liver sections.
Real time polymerase chain reaction (PCR) quantification of mRNA
RNA was isolated from liver, duodenum, and spleen using the RNeasy mini kit (Qiagen, Clifton Hill, Australia) according to the manufacturer’s instructions. Any contaminating genomic DNA was removed by treatment with RQ1 RNase free DNaseI (Promega, Annandale, Australia). RNA (1–2 μg) was reverse transcribed using Superscript III (Invitrogen, Mulgrave, Australia) according to the manufacturer’s instructions. Real time reaction mixes contained 2 μl of cDNA (diluted 1:10 or 1:20), 200 nM of each primer (table 1 ▶), and SYBR green PCR master mix (Applied Biosystems, Warrington, UK). Reactions were performed on the Rotorgene RG3000 (Corbett Research, Brisbane, Australia) using the following conditions: two minutes denature at 95°C followed by 50 cycles of 95°C for 30 seconds, 55°C for 30 seconds, and 72°C for 30 seconds. Fluorescence due to SYBR green binding to double stranded DNA was measured during the extension steps. Melt curve analysis was routinely performed to monitor primer dimer levels by raising the temperature from 50°C to 99°C at 1°C/minute while monitoring fluorescence continuously. All experiments were performed in duplicate, and a single batch of cDNA was used for all genes analysed. A standard curve was performed for each run comprising serial dilutions of pooled cDNA to ensure the integrity of the results. Expression of all target genes was determined by normalising to the respective β-actin levels.
Table 1.
Real time polymerase chain reaction primers
| Gene | Sequence (5′-3′) | Reference |
| β-actin | F: GAC GGC CAA GTC ATC ACT ATT G | Dupic24 |
| R: CCA CAG GAT TCC ATA CCC AAG A | ||
| Ferroportin 1 | F: TTG CAG GAG TCA TTG CTG CTA | Dupic24 |
| R: TGG AGT TCT GCA CAC CAT TGA T | ||
| Hfe | F: CTG AAA GGG TGG GAC TAC ATG TTC | Dupic24 |
| R: GGA CAC CAC TCC CAA CTT CGT | ||
| TfR1 | F: CAT GAG GGA AAT CAA TGA TCG TA | Dupic24 |
| R: GCC CCA GAA GAT ATG TCG GAA | ||
| DMT1 | F: GGC TTT CTT ATG AGC ATT GCC TA | Dupic24 |
| R: GGA GCA CCC AGA GCA GCT TA | ||
| DcytB | F: GCA GCG GGC TCG AGT TTA | Dupic24 |
| R: TTC CAG GTC CAT GGC AGT CT | ||
| Hephaestin | F: TTG TCT CAT GAA GAA CAT TAC AGC AC | Dupic24 |
| R: CAT ATG GCA ATC AAA GCA GAA GA | ||
| TfR2 | F: CTA TCT GGT CCT GAT CAC CCT | |
| R: TCA GGG TTG ACA TCT TCA TCG A | ||
| Total hepcidin | F: AGA GCT GCA GCC TTT GCA C* | |
| R: GAG GTC AGG ATG TGG CTC TA | ||
| Hepcidin1 | F: AGA GCT GCA GCC TTT GCA C* | |
| R: ACA CTG GGA ATT GTT ACA GCA TTT A | ||
| Hepcidin2 | F: AGA GCT GCA GCC TTT GCA C* | |
| R: GGA GGG TTT GTT ACA GCA CTG | ||
| Hemojuvelin | F: ATG CCG TGT CCA AGG AGC TT | |
| R: TCC ACC TCA GCC TGG TAG AC |
*The same forward (F) primer was used for total hepcidin, hepcidin 1, and hepcidin 2.
R, reverse; DMT1, divalent metal transporter 1; DcytB, duodenal cytochrome B; TfR2, transferrin receptor 2.
Western blotting
Liver homogenates (100 μg) were electrophoresed on either 10% Tris-glycine sodium dodecyl sulphate-polyacrylamide gels or on 4–12% Novex Bis-Tris gradient gels with a MES buffering system (Invitrogen, Mulgrave, Australia), transferred onto Hybond-C+ or PVDF membrane, and blocked in 10% skim milk powder and 0.5% Tween 20 in phosphate buffered saline (PBS) (blocking buffer) overnight at 4°C. Anti-TfR2 (1 μg/ml), anti-TfR1 (1 μg/,ml), antiferritin (1:200), antiprohepcidin (2 μg/ml) or antiactin (1:400) was diluted in blocking buffer and applied to the blots for one hour at room temperature. Blots were washed extensively with 0.1% Tween 20 in PBS and then incubated with antirabbit, antimouse, or antisheep horseradish peroxidase for one hour at room temperature. Blots were washed and Supersignal West Pico chemiluminescent substrate (Pierce Chemical Co., Rockford, Illinois, USA) was applied to the blot and exposed to film. Protein expression was quantified by densitometry and normalised to levels of β-actin.
Immunohistochemistry
Portions of liver were placed in 10% buffered formalin for two hours, transferred into 70% ethanol, processed, and embedded in paraffin. Tissue sections (3 μm) were subjected to citrate buffer antigen retrieval and blocked with 10% goat serum for 30 minutes. Sections were incubated with anti-TfR2 (10 μg/ml) and antiprohepcidin (10 μg/ml) antibodies at 4°C in the dark overnight. Slides were rinsed with PBS, incubated with biotinylated goat antirabbit antibody (Dako A/S, Denmark) for one hour, rinsed briefly in PBS, and incubated with StreptABComplex/horseradish peroxidase for 30 minutes. After rinsing with PBS the slides were incubated with Sigma Fast 3,3′-diaminobenzidine tablets in PBS for four minutes. The slides were counterstained with Harris haematoxylin, dehydrated through alcohol series and xylene, and mounted with DePeX mounting medium. Images were taken using an Olympus DPII digital camera.
Statistics
Variables were compared between groups using both parametric (one way ANOVA and the Student’s t test) and non-parametric (Kruskal-Wallis and Mann-Whitney) statistical tests using the SPSS software package. Correlations were performed between variables using Microsoft Excel. A p value <0.05 was considered statistically significant.
RESULTS
Assessment of iron status
Measurement of iron indices in TfR2-KO mice showed that they developed significant iron overload by 10 weeks of age. Transferrin saturation was significantly elevated (83.7 v 36.7%; p<0.001) in homozygous TfR2-KO mice compared with wild-type and heterozygous mice (fig 1 ▶). Hepatic iron concentration was approximately fivefold higher in homozygous TfR2-KO mice (1023.6 v 188.8 μg/g; p<0.001). There was no significant difference in transferrin saturation or hepatic and splenic iron concentrations between wild-type and heterozygous mice. Homozygous TfR2-KO mice had significantly less splenic iron than wild-type and heterozygous mice (fig 1 ▶).
Figure 1.

Measurement of iron indices. Mean values for transferrin saturation, hepatic iron concentration, and splenic iron concentration are shown for 10 week old wild-type, heterozygous, and homozygous transferrin receptor 2-knockout mice. Error bars denote SDs. **p<0.01.
The pattern of hepatic iron loading in TfR2-KO mice was assessed by Perls’ staining of liver sections. Iron accumulation was clearly visible in hepatocytes with a gradient from periportal to pericentral regions (fig 2 ▶), a pattern typical of that seen in patients with TfR2 associated haemochromatosis25 and in mice with mutant TfR2.7 No staining for iron was detected in wild-type or heterozygous mice.
Figure 2.

Perls’ staining of liver sections. Liver sections from 10 week old wild-type (A), heterozygous (B), and homozygous (C) transferrin receptor 2-knockout mice stained with Perls’ Prussian blue for iron. Original magnification 20×.
Expression of iron related genes in the liver, duodenum, and spleen
Levels of iron related mRNAs were measured in the livers of homozygous TfR2-KO, heterozygous, and wild-type mice using real time PCR. TfR2 mRNA was absent in the liver of homozygous mice and levels in heterozygous mice were approximately half those of wild-type mice (table 2 ▶). TfR1 and divalent metal transporter 1 (DMT1) mRNA levels were significantly reduced (eightfold and 1.5-fold, respectively) in homozygous mice compared with wild-type and heterozygous mice (table 2 ▶). Hepcidin 2 mRNA levels were significantly higher in homozygous mice (p = 0.016). However, levels of total hepcidin and hepcidin 1 mRNA were similar in all three groups and there was a close correlation between the two (r = 0.92; p = 2.17×10−13). Hepcidin 2 mRNA levels did not correlate with either total hepcidin or hepcidin 1 mRNA, suggesting that hepcidin 1 accounts for most of the total hepcidin and is the predominant form produced in the liver. This agrees with previously published data in C57BL/6J mice where hepcidin 1 was found to be the predominant form.26
Table 2.
Quantification of mRNA transcripts in the liver, duodenum, and spleen
| Tissue | mRNA | Wild-type | Heterozygous TfR2-KO | Homozygous TfR2-KO |
| Liver | TfR2 | 0.1887 (0.0868) | 0.0989* (0.0453) | 0.0000** (0.0000) |
| TfR1 | 0.00464 (0.00359) | 0.00421 (0.00270) | 0.00053** (0.00050) | |
| Total hepcidin | 67.00 (61.09) | 81.16 (45.58) | 59.06 (41.30) | |
| Hepcidin1 | 19.50 (14.55) | 20.47 (12.76) | 12.90 (10.11) | |
| Hepcidin2 | 3.049 (3.542) | 4.551 (4.095) | 9.634* (6.071) | |
| Hfe | 0.0665 (0.0187) | 0.0707 (0.0301) | 0.0496 [0.0410**] (0.0290 [0.0103]) | |
| Fpn1 | 0.0245 (0.0100) | 0.0251 (0.0146) | 0.0287 [0.0187*] (0.0318 [0.0030]) | |
| Hjv | 0.0145 (0.0020) | 0.0188 (0.0086) | 0.0178 [0.0105**] (0.0234 [0.0026]) | |
| DMT1 | 0.0580 (0.0396) | 0.0580 (0.0252) | 0.0384* (0.0134) | |
| Duodenum | Fpn1 | 0.1060 (0.0304) | 0.0831 (0.0501) | 0.0972 (0.0512) |
| DcytB | 0.0827 (0.0636) | 0.0942 (0.1134) | 0.1541 (0.1610) | |
| TfR1 | 0.00339 (0.00203) | 0.00324 (0.00200) | 0.00318 (0.00167) | |
| DMT1 | 0.00393 (0.00340) | 0.00386 (0.00469) | 0.00427 (0.00219) | |
| Hephaestin | 0.001535 (0.000910) | 0.000865 (0.000590) | 0.000815 (0.000501) | |
| Spleen | Fpn1 | 0.0221 (0.0156) | ND | 0.0158 (0.0101) |
| TfR2 | 0.00122 (0.00090) | ND | 0.00000* (0.00000) | |
| TfR1 | 0.00807 (0.00474) | ND | 0.00338 (0.00370) | |
| Total hepcidin | 0.00357 (0.00493) | ND | 0.00213 (0.00163) | |
| DMT1 | 0.00154 (0.00075) | ND | 0.00208 (0.00338) |
Mean (SD) values are shown, normalised to β-actin mRNA levels.
[Outliers removed from analysis]. ND, not done.
*p<0.05; **p<0.01.
DcytB, duodenal cytochrome B; DMT1, divalent metal transporter 1; Fpn1, ferroportin; Hjv, hemojuvelin; KO, knockout; TfR1, transferrin receptor 1; TfR2, transferrin receptor 2.
Levels of Hfe, Hjv, and ferroportin1 (Fpn1) mRNA in the liver were similar in all groups and there was no significant difference when compared using the Student’s t test. However, the non-parametric Mann-Whitney test suggested there was significantly less Hfe and Hjv mRNA in the livers of homozygous TfR2-KO mice compared with wild-type and heterozygous mice (p = 0.035 and 0.007, respectively). On further inspection we noted that there was one mouse with much higher levels of Hfe, Hjv, and Fpn1 in the homozygous group. When this mouse, or outlier, was removed from the analysis the Student’s t test also delivered a significant result for both Hfe and Hjv (p<0.001 and p = 0.001, respectively), with Hfe being 1.7- and Hjv being 1.6-fold lower in the homozygous group (table 2 ▶). The Fpn1 level was also reduced 1.3-fold in the homozygous group compared with wild-type and heterozygous mice (table 2 ▶) but this was only marginally significant when using the Student’s t test (p = 0.043) and not significant when using other statistical tests (one way ANOVA, Kruskal-Wallis, and Mann-Whitney).
Levels of iron related mRNAs in the duodenum were similar in all three groups of mice and there was no significant difference in expression of these molecules (table 2 ▶). TfR2 mRNA in the duodenum was virtually undetectable in all three groups. Levels of iron related mRNAs in the spleen were compared between wild-type and homozygous mice only (table 2 ▶). TfR2 mRNA was undetectable in the spleens of homozygous TfR2-KO mice. Wild-type mice had approximately 150-fold less TfR2 in the spleen compared with the liver. Levels of all other mRNAs in the spleen were similar between the two groups, except that there was less TfR1 in homozygous mice. This approached significance when compared using the Student’s t test (p = 0.054) and was significant when using the Mann-Whitney test (p = 0.013).
Hepatic protein expression in the liver
Immunoblotting was used to determine expression of key iron related proteins in the liver. Levels of TfR2 protein were determined using a TfR2 specific antibody (fig 3A ▶) and correlated well with levels of TfR2 mRNA (r = 0.88, p<0.001). A representative immunoblot of the key iron related proteins is shown in fig 3B ▶. TfR2 protein was completely absent in the livers of TfR2-KO mice. Heterozygous mice had intermediate levels compared with wild-type mice. These results were confirmed using an antibody raised against the extracellular domain of TfR2 and show that any truncated protein lacking exons 2–6 is not produced in knockout mice (data not shown). TfR1 protein levels were significantly decreased (3.9-fold, p<0.001) while hepatic ferritin levels were significantly raised (4.5-fold, p<0.001) in homozygous mice compared with wild-type and heterozygous mice (fig 3 ▶), consistent with hepatic iron loading. Levels of hepatic prohepcidin protein varied between animals but there was no significant difference in expression between groups of mice, despite iron loading and increased transferrin saturation in TfR2-KO mice. The prohepcidin antibody used detects both mouse prohepcidin 1 and prohepcidin 2 (unpublished observations). Levels of hepatic prohepcidin protein correlated with total hepcidin (r = 0.39, p = 0.031) and hepcidin 1 mRNA (r = 0.42, p = 0.020) but not hepcidin 2 mRNA, further confirming that hepcidin 1 is the predominant form produced in mouse liver.
Figure 3.

Quantification of hepatic protein levels. (A) Western blot analysis of 100 μg of mouse liver (lane 1) and kidney (lane 2) homogenates with antibodies against transferrin receptor 2 (TfR2), transferrin receptor 1 (TfR1), and the Golgi marker GS28 as a loading control. Antibodies were specific for TfR2. (B) Total liver protein from 10 week old mice was western blotted with antibodies against TfR2, TfR1, ferritin, prohepcidin, and β-actin. Representative western blot of three mice: lane 1, wild-type; lane 2, heterozygous; lane 3, homozygous TfR2-knockout (KO). (C) Quantification of protein levels after normalising to β-actin. Mean (SD) values are shown for wild-type, heterozygous, and homozygous TfR2-KO mice. *p<0.05; **p<0.01.
Immunohistochemistry of TfR2 and prohepcidin in the liver
TfR2 and prohepcidin expression in the livers of homozygous TfR2-KO, heterozygous, and wild-type mice was analysed by immunohistochemistry. Three mice from each group were selected for analysis. Staining was similar within each group and typical images are shown in figs 4 ▶ and 5 ▶. No TfR2 staining was detected in the liver of homozygous TfR2-KO mice. In heterozygous and wild-type mice, TfR2 was expressed in hepatocytes and localised to the cell surface, predominantly on the basolateral surface of the plasma membrane (fig 4 ▶) with a faint signal observed on the apical surface. Prohepcidin was detected in the livers of all mice consistent with the immunoblotting results. Prohepcidin staining had a punctate “beaded” expression pattern reminiscent of the Golgi apparatus and secretory vesicles (fig 5 ▶).
Figure 4.
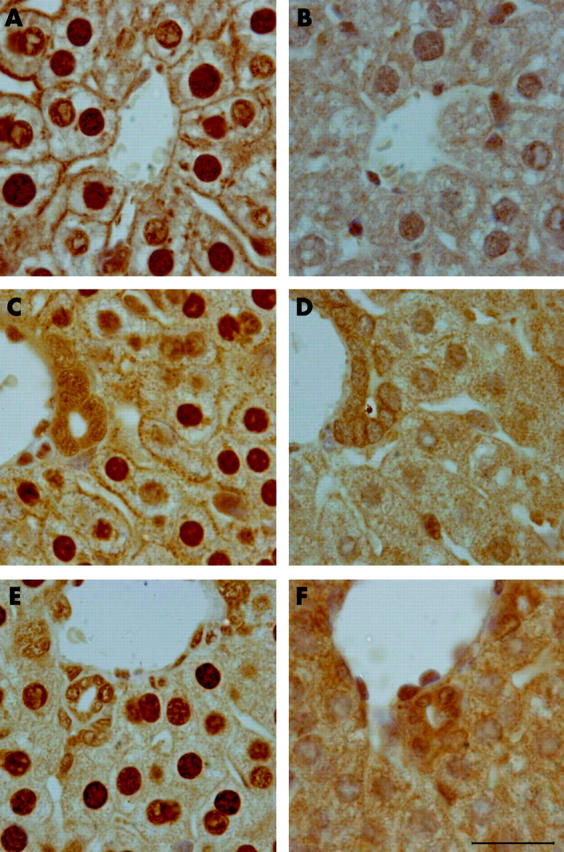
Immunohistochemical localisation of transferrin receptor 2 (TfR2) in the liver. Liver sections from 10 week old wild-type (A,B), heterozygous (C, D), and homozygous (E, F) TfR2-knockout mice were incubated with anti-TfR2 antibodies (A, C, E) or control rabbit IgG (B, D, F). Original magnification 100×. Scale bar 200 μm.
Figure 5.
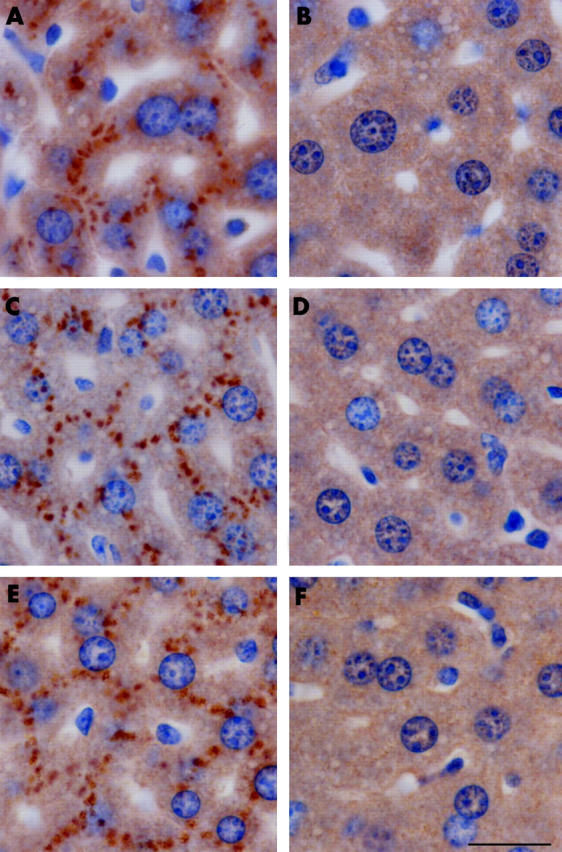
Immunohistochemical localisation of prohepcidin in the liver. Liver sections from 10 week old wild-type (A, B), heterozygous (C, D), and homozygous (E, F) transferrin receptor 2-knockout mice were incubated with antiprohepcidin antibodies (A, C, E) or control rabbit IgG (B, D, F). Original magnification 100×. Scale bar 200 μm.
DISCUSSION
In this report we have described the phenotype and analysed expression of iron related molecules in the liver, duodenum, and spleen of mice with complete knockout of the TfR2 gene. These mice develop parenchymal iron overload and features of TfR2 associated haemochromatosis. There is an increase in transferrin saturation which is accompanied by hepatic iron loading and splenic iron deficiency. In these mice there is no upregulation of hepcidin mRNA or protein expression, as would be expected in mice with high iron stores, and there is slight downregulation of Hfe and Hjv mRNA.
TfR2-KO mice have an iron overload phenotype which is similar to TfR2-Y245X mutant mice and to patients with TfR2 associated haemochromatosis. Homozygous TfR2-KO mice have increased transferrin saturation compared with both wild-type and heterozygous mice. At 10 weeks of age, homozygous mice have approximately fivefold more iron in the liver compared with wild-type and heterozygous mice. This is reflected in an approximate fivefold increase in hepatic ferritin levels. The spleens of TfR2-KO mice are relatively iron deficient. Splenic iron deficiency has also been observed in TfR2-Y245X mice7 and Hfe-KO mice, and is consistent with the iron deficiency of the macrophage compartment in patients with hereditary haemochromatosis.
The phenotypes of TfR2 and HFE associated haemochromatosis are very similar,25 suggesting that both TfR2 and HFE either function in the same pathway or in parallel pathways regulating iron homeostasis. It now seems apparent that the final regulatory target of these molecules is the liver expressed peptide hepcidin. In HFE associated haemochromatosis and Hfe-KO mice there is no upregulation of hepcidin despite increased iron stores.14,15 While this manuscript was in preparation, two studies have also implicated TfR2 in the iron induced hepcidin pathway.27,28 Urinary hepcidin levels were low in patients with TfR2 associated haemochromatosis and hepcidin mRNA was not upregulated in mice homozygous for the TfR2-Y245X mutation. Hepcidin functions by inhibiting iron release from cells. A recent study has shown that hepcidin achieves this by binding to the iron exporter ferroportin 1 on the cell surface and inducing its internalisation.29
In order to examine the effect of complete absence of TfR2, we studied expression levels of iron related molecules in the liver, duodenum, and spleen. While TfR2-KO mice had no expression of TfR2 mRNA or protein in the liver, heterozygous mice had intermediate levels of both TfR2 mRNA and protein. Low TfR1 and DMT1 mRNA levels together with increased levels of ferritin all point to reduced IRP binding activity and iron overload within hepatocytes. We did not see an increase in Fpn1 mRNA in TfR2-KO mice and in fact there was a slight decrease when an outlier was removed from the analysis. This may indicate transcriptional downregulation of Fpn1 in the livers of TfR2-KO mice. The level of Fpn1 protein was not measured and therefore the effect of iron overload on IRE-IRP mediated regulation of Fpn1 protein could not be assessed.
We have shown that expression of hepcidin mRNA in the liver is not consistent with the observed hepatic iron loading in TfR2-KO mice. Total hepcidin and hepcidin 1 mRNA levels were not increased in homozygous TfR2-KO mice despite iron loading. This is consistent with the observed loss of hepcidin mRNA regulation in mice with the TfR2-Y245X mutation.28 We did see a significant increase in hepcidin 2 mRNA in homozygous TfR2-KO mice, suggesting that there may be some intact regulation specifically of hepcidin 2 in response to iron loading. However, levels of hepcidin 2 in the liver are much lower than hepcidin 1 and there was no effect of this increase on total hepcidin mRNA or prohepcidin protein. Humans only have one hepcidin gene and therefore the significance of this finding in the human situation is not clear. Also, another study concluded that hepcidin 2 in mice was not functional in iron metabolism and the effect of hepcidin on iron metabolism was mediated only by hepcidin 1.30 In addition to hepcidin mRNA, we studied expression of prohepcidin protein by immunoblotting. Levels of prohepcidin protein did not increase in TfR2-KO mice (fig 3 ▶). This is the first analysis of prohepcidin protein levels in mice with defective TfR2 expression.
In addition to hepcidin mRNA and protein levels, we analysed expression of other iron related mRNAs in the liver. On closer inspection of Hfe and Hjv mRNA expression, a small but statistically significant reduction in these molecules in TfR2-KO mice was detected. This difference was observed using non-parametric tests and parametric tests after removing an outlier from the analysis. It is not clear at present whether this reduction in expression is due to iron loading of the liver or a direct consequence of the loss of TfR2. Whether the decrease in Hfe and Hjv in TfR2-KO mice is related to the loss of hepcidin regulation is also not clear. However, it appears that TfR2, Hfe, and Hjv are all related in the iron regulated pathway leading to hepcidin expression.
There was very little difference in expression of iron related mRNAs in the duodenum and spleen of TfR2-KO mice. TfR2 mRNA was undetectable in the duodenum of all mice. In the duodenum, mRNAs encoding the iron transporters DMT1 and Fpn1 and facilitators DcytB and hephaestin were not downregulated despite iron loading. Contrary to this study, Kawabata et al observed an increase in duodenal DMT1 mRNA levels in TfR2-Y245X mice, suggesting that duodenal iron absorption was increased.28 This difference may be due to the age of the mice studied (four months v 10 weeks). Also, Kawabata et al specifically measured levels of the exon 1A form of DMT1 whereas in this study total DMT1 mRNA was measured. An increase in duodenal iron transporters would not necessarily be expected in TfR2-KO mice as the loss of hepcidin regulation in the liver of homozygous TfR2-KO mice leads to hepcidin levels comparable with wild-type mice. Even though there was no observed increase in duodenal iron transporters, iron absorption would still be increased relative to iron stores. In the spleen, levels of TfR2 were approximately 150-fold lower. These results confirm that the primary location of TfR2 is the liver. In the spleen, we observed a reduction in TfR1 mRNA in homozygous TfR2-KO mice. This is contrary to what might be expected; low iron levels in the cell would normally cause stabilisation of TfR1 mRNA by IRE-IRP mediated mechanisms. Therefore, rather than the iron status of the cell determining expression of TfR1 in the spleen, the low splenic iron concentration in TfR2-KO mice may be a consequence of downregulation of TfR1. Mechanisms leading to downregulation of TfR1 in the spleen of these mice are not clear and require further study.
Using immunohistochemistry, we confirmed that TfR2 is absent from the liver of TfR2-KO mice. TfR2 localises mainly to the cell surface of hepatocytes, predominantly at the basolateral surface in both wild-type and heterozygous mice, with some staining also detected on the apical surface. The distribution of TfR2 at the plasma membrane of the hepatocyte is consistent with its proposed function. TfR2 could be acting as a sampler of transferrin, perhaps sensing either the iron saturation of transferrin or the amount of diferric transferrin in blood. Activation of TfR2 by transferrin could then cause a signalling cascade resulting in induction of hepcidin mRNA expression.
The immunohistochemical localisation of prohepcidin in the liver was the same in homozygous TfR2-KO, heterozygous, and wild-type mice. Prohepcidin has a “beaded” expression pattern which is reminiscent of the Golgi and secretory pathway that we have previously observed in other mouse strains.22 Therefore, in TfR2-KO mice there appears to be no upregulation of hepcidin in response to iron loading and no post-transcriptional regulation of hepcidin protein.
In conclusion, we have shown that mice with complete absence of TfR2 develop haemochromatosis. TfR2 is required for the iron regulated expression of hepcidin. The pathway in which TfR2 functions is likely to involve Hfe and Hjv. While our results cannot definitively place these proteins in precise positions within a pathway, they suggest that TfR2 is upstream of both Hfe and Hjv in a signalling cascade which results in induction of hepcidin expression. These results support the concept of the liver as the central regulator of body iron homeostasis.
Acknowledgments
This work was supported in part by grants from the National Health and Medical Research Council of Australia (953219) and the National Institutes of Health, USA (5R01DK057648–04).
Abbreviations
DcytB, duodenal cytochrome B
DMT1, divalent metal transporter 1
Fpn1, ferroportin
Hjv, hemojuvelin
KO, knockout
TfR1, transferrin receptor 1
TfR2, transferrin receptor 2
PCR, polymerase chain reaction
PBS, phosphate buffered saline
Conflict of interest: None declared.
REFERENCES
- 1.Kawabata H, Yang R, Hirama T, et al. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J Biol Chem 1999;274:20826–32. [DOI] [PubMed] [Google Scholar]
- 2.Fleming RE, Migas MC, Holden CC, et al. Transferrin receptor 2: continued expression in mouse liver in the face of iron overload and in hereditary hemochromatosis. Proc Natl Acad Sci U S A 2000;97:2214–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawabata H, Germain RS, Vuong PT, et al. Transferrin receptor 2-alpha supports cell growth both in iron-chelated cultured cells and in vivo. J Biol Chem 2000;275:16618–25. [DOI] [PubMed] [Google Scholar]
- 4.West AP Jr, Bennett MJ, Sellers VM, et al. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J Biol Chem 2000;275:38135–8. [DOI] [PubMed] [Google Scholar]
- 5.Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet 2000;25:14–15. [DOI] [PubMed] [Google Scholar]
- 6.Roetto A, Daraio F, Alberti F, et al. Hemochromatosis due to mutations in transferrin receptor 2. Blood Cells Mol Dis 2002;29:465–70. [DOI] [PubMed] [Google Scholar]
- 7.Fleming RE, Ahmann JR, Migas MC, et al. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc Natl Acad Sci U S A 2002;99:10653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krause A, Neitz S, Magert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett 2000;480:147–50. [DOI] [PubMed] [Google Scholar]
- 9.Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 2001;276:7806–10. [DOI] [PubMed] [Google Scholar]
- 10.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem 2001;276:7811–19. [DOI] [PubMed] [Google Scholar]
- 11.Nicolas G, Chauvet C, Viatte L, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest 2002;110:1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci U S A 2001;98:8780–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet 2003;33:21–2. [DOI] [PubMed] [Google Scholar]
- 14.Bridle KR, Frazer DM, Wilkins SJ, et al. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet 2003;361:669–73. [DOI] [PubMed] [Google Scholar]
- 15.Ahmad KA, Ahmann JR, Migas MC, et al. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol Dis 2002;29:361–6. [DOI] [PubMed] [Google Scholar]
- 16.Roy CN, Custodio AO, de Graaf J, et al. An Hfe-dependent pathway mediates hyposideremia in response to lipopolysaccharide-induced inflammation in mice. Nat Genet 2004;36:481–5. [DOI] [PubMed] [Google Scholar]
- 17.Frazer DM, Wilkins SJ, Millard KN, et al. Increased hepcidin expression and hypoferraemia associated with an acute phase response are not affected by inactivation of HFE. Br J Haematol 2004;126:434–6. [DOI] [PubMed] [Google Scholar]
- 18.Lee P, Peng H, Gelbart T, et al. The IL-6- and lipopolysaccharide-induced transcription of hepcidin in HFE-, transferrin receptor 2-, and beta 2-microglobulin-deficient hepatocytes. Proc Natl Acad Sci U S A 2004;101:9263–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papanikolaou G, Samuels ME, Ludwig EH, et al. Mutations in HFE2 cause iron overload in chromosome 1q-linked juvenile hemochromatosis. Nat Genet 2004;36:77–82. [DOI] [PubMed] [Google Scholar]
- 20.Levy JE, Montross LK, Cohen DE, et al. The C282Y mutation causing hereditary hemochromatosis does not produce a null allele. Blood 1999;94:9–11. [PubMed] [Google Scholar]
- 21.Wallace DF, Tonks ID, Zournazi A, et al. Inactivation of the murine transferrin receptor 2 gene using the Cre recombinase:loxP system. Genesis 2004;39:38–41. [DOI] [PubMed] [Google Scholar]
- 22.Wallace DF, Summerville L, Lusby PE, et al. Prohepcidin localises to the Golgi compartment and secretory pathway in hepatocytes. J Hepatol 2005; (in press). [DOI] [PubMed]
- 23.Torrance JD, Bothwell TH. A simple technique for measuring storage iron concentrations in formalinised liver samples. S Afr J Med Sci 1968;33:9–11. [PubMed] [Google Scholar]
- 24.Dupic F, Fruchon S, Bensaid M, et al. Duodenal mRNA expression of iron related genes in response to iron loading and iron deficiency in four strains of mice. Gut 2002;51:648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pietrangelo A. Non-HFE hemochromatosis. Hepatology 2004;39:21–9. [DOI] [PubMed] [Google Scholar]
- 26.Courselaud B, Troadec MB, Fruchon S, et al. Strain and gender modulate hepatic hepcidin 1 and 2 mRNA expression in mice. Blood Cells Mol Dis 2004;32:283–9. [DOI] [PubMed] [Google Scholar]
- 27.Nemeth E, Roetto A, Garozzo G, et al. Hepcidin is decreased in TFR2 hemochromatosis. Blood 2005;105:1803–6. [DOI] [PubMed] [Google Scholar]
- 28.Kawabata H, Fleming RE, Gui D, et al. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 2005;105:376–81. [DOI] [PubMed] [Google Scholar]
- 29.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004;306:2090–3. [DOI] [PubMed] [Google Scholar]
- 30.Lou DQ, Nicolas G, Lesbordes JC, et al. Functional differences between hepcidin 1 and 2 in transgenic mice. Blood 2004;103:2816–21. [DOI] [PubMed] [Google Scholar]