

Abstract
Background and aims: CREB (cAMP response element binding protein) transcription factors are key regulators of homeostatic functions in the liver, and CRE binding is increased in hepatic inflammation. During chronic hepatitis B virus (HBV) infection, mutations or deletions in the pre-S region are frequently observed. These mutations can affect the pre-S2/S promoter controlling HBV envelope protein expression (hepatitis B surface antigen (HBsAg)) and have been associated with worsened clinical outcome. We aimed to test if CREB activation impacts on HBsAg expression.
Methods: The effect of the CREB inducer protein kinase A (PKA) was tested by coexpression with HBV wild-type vector in vitro. Luciferase reporter gene constructs were cloned to identify novel regulatory regions for the HBV pre-S2/S promoter. Electrophoretic mobility shift assay (EMSA) gelshift and supershift experiments were conducted to confirm DNA transcription factor binding.
Results: Coexpression of HBV and PKA resulted in HBV-S mRNA induction and enhanced small envelope protein expression. We identified a CREB binding motif in the transcribed part of the pre-S2 region, contributing to basal S promoter activity via binding of activating transcription factor 2 (ATF2). A second CREB motif closely linked to the S-ATG showed a similar binding pattern involving ATF2 and CREB1, without appearing essential for basal promoter activity. Moreover, a sequence in the pre-S2 region is responsible for further transcriptional induction via CREB activators such as PKA and forskolin. EMSA experiments indicate that CREB1 and ATF4 are involved in complex formation conferring PKA dependent promoter activation.
Conclusions: Our data suggest a novel mechanism by which HBV may utilise CREB/PKA signal transduction pathways of hepatocytes to enhance its HBsAg expression during homeostasis and hepatic inflammation.
Keywords: cAMP response element, cAMP response element binding protein, protein kinase A, hepatitis B virus, promoter
The cyclic AMP response element (CRE) has been implicated in the regulation of gene expression and cellular processes important in hepatocyte function, such as gluconeogenesis, lipid metabolism, and proliferation.1–3 Recently, increased CRE binding has been shown in response to proinflammatory cytokines in vitro as well as to lipopolysaccharide injection in rats.4 This suggests that transcription factors of the CREB (cAMP response element binding protein) family do not only regulate homeostatic functions in the liver but also mediate cellular responses to hepatic inflammation. CREB factors are normally activated by phosphorylation (for example, by protein kinase A (PKA)).5,6
Persistent infection with the hepatitis B virus (HBV) represents a major health problem worldwide, with more than 350 million chronically infected patients at risk of developing liver cirrhosis or hepatocellular carcinoma. Natural occurring mutations are very common during chronic infection, and the highest heterogenicity in the HBV genome is found in the open reading frame (ORF) for the envelope protein (S-ORF).7 S-ORF is organised into the pre-S1, pre-S2, and S regions under the transcriptional control of two promoters (pre-S1, pre-S2/S). The highest rate of mutations in the HBV genome can be found in the pre-S1 and pre-S2 regions where point mutations, deletions, or genetic recombinations occur.7,8 We and others have reported that pre-S mutations can be associated with a more severe course of hepatitis and clinical complications such as cholestating hepatitis, liver cirrhosis, or hepatocellular carcinoma.9–16 Some of these mutations can be attributed to altered function of the mutated envelope proteins9,17–19 but many affect pre-S2/S promoter activity (“S promoter”) controlling the expression of the middle and small (hepatitis B surface antigen (HBsAg)) envelope proteins.
The pre-S1 promoter is located upstream from the first ATG found in S-ORF and regulates transcription of the 2.4 kb S mRNA, which encodes for the large surface protein of HBV (LHBs). The pre-S1 promoter consists of a TATA box, has a relatively low activity, and is regulated by at least two liver enriched transcription factors, HNF-1 and HNF-3.8 In contrast, the TATA-less pre-S2/S promoter (S promoter) confers the strongest promoter activity in comparison with other HBV promoters and controls transcription of the 2.1 kb S mRNA generating the middle (MHBs) and small (SHBs) hepatitis B surface proteins. A single upstream CCAAT element has been identified as essential for high level expression. Moreover, four functional SP1 sites as well as a binding site for the transcription factor NF-1 also contribute to promoter activity.20–24
The three envelope proteins have distinct functions. LHBs has been shown to be involved in hepatocyte entry25,26 whereas MHBs appears dispensable for virion formation.27,28 SHBs, historically referred to as HBsAg, are expressed at extremely high levels. They are not only essential for virion formation and part of the mature virion, but are also released as spheric or filamentous subviral particles alone.29 SHBs carry the most important antigenic epitope in the immunodominant “a-determinant” loop, and excess release of HBsAg particles has been associated with viral persistence as it may allow escape of the humoral anti-HBs response.30
In this study, our aim was to test if CREB transcription factors enhance HBs expression. Coexpression of HBV wild-type plasmid and the CREB activator PKA increased HBs transcription and protein expression in vitro. Further analysis led to characterisation of CREB binding motifs in the transcribed pre-S2 region supporting basal pre-S2/S promoter activity by binding of CREB1/activating transcription factor 2 (ATF2) and of an Ets-like binding motif mediating transcriptional activation to CREB and its activators PKA and forskolin.
METHODS
Cell culture and transfection experiments
Human Huh7 hepatoma cells, shown to be negative for any HBV marker, were cultured in Dulbeccos’s modified Eagle’s medium supplemented with 10% fetal bovine serum under 5% CO2 at 37°C. DNA was transfected using the calcium phosphate coprecipitation method.20 To test the potential effect of CREB/PKA on HBV-S expression, cotransfection experiments of 3 µg of replication competent HBV wild-type 1.28 plasmid (genotype A, subtype adw2) and increasing concentrations of PKA expression plasmid were performed.31 pBluescript (pBS; Stratagene, La Jolla, California, USA) was added to keep the amount of transfected DNA constant at 5 µg. For reporter gene experiments, cells were transfected with 2.5 µg of the luciferase construct, 2 µg pBS, and 0.5 µg of pCMV β-galactosidase20 as an internal standard.
Northern and western blot analysis
Total RNA from Huh7 cells was isolated using the RNeasy Kit (Qiagen, Hilden, Germany).32 Northern blot analysis was performed as previously described using an α32P-radiolabelled 2.5 kb monomeric HBV probe that was capable of detecting all HBV mRNAs.33 Data were quantified using the NIH imager system and normalised to the applied total RNA (18S signal).34 For protein analysis, whole cellular protein extracts were collected 48 and 72 hours after transfection, separated on a 10% sodium dodecyl sulphate-polyacrylamide gel and blotted onto a nitrocellulose membrane, as previously described.34 Membranes were then probed with the monoclonal mouse antihuman HBsAg antibody 3E7 (#M3506; Dako, Carpinteria, California, USA). As secondary antibody, anti-mouse IgG (#AQ127P; Chemicon International, Temecula, California, USA) was used. Reblotting of the same membranes with an anti-α-tubulin antibody (#sc-8035; Santa Cruz Biotechnology, Santa Cruz, California, USA) was employed to demonstrate equal protein loading. The antigen-antibody complexes were visualised using the ECL-chemiluminescence kit (Amersham, Arlington Heights, Illinois, USA).
Cloning of luciferase expression plasmids, luciferase, and beta-galactosidase reporter assays
A computer based search for potential transcription factor binding sites was performed using MatInspector software.35 For construction of luciferase reporter plasmids, predicted promoter sequences were amplified by polymerase chain reaction (PCR) (Hotstart Herculase; Stratagene) using the HBV 1.28 wild-type plasmid as a template.36 Oligonucleotides used for PCR (table 1 ▶) were purchased from MWG Biotech (Ebersberg, Germany) and flanked by XhoI and SacI restriction sites, respectively.
Table 1.
Primer sequences used for generation of luciferase expression constructs
| Sense primer | 5′ – CAGATTGGGACTTCAACCCCGTC – 3′ |
| 1–as | 5′ - CGCTGCCTCGAGGTTCGTCACAGGGTCCCCAGT – 3′ |
| 2–as | 5′ - TACTCGAGGAGGCAATATTCGGAGC – 3′ |
| 3–as | 5′ - TACTCGAGGACTCTGGGATCCTGC – 3′ |
| 4–as | 5′ - TACTCGAGGGTGGAAGGCAGTGG– 3′ |
| 5–as | 5′ - CCACTGCATGGCTCGAGGATGACTG – 3′ |
| 1B–as | 5′ - TACTCGAGCAGTCCTCGCGGAGATTGACGA – 3′ |
| 1Bmut–as | 5′ - TACTCGAGCAGTCCTCGCGGAGATTCCCGA – 3′ |
| 1 mut/wt–as | 5′ - TACTCGAGGTTCGTCACAGGGTCCCCAGTCCTCGCGGAGATTCCCGAG – 3′ |
| 1 wt/mut–as | 5′ - TACTCGAGGTTCGGGGCAGGGTCCCCAGTCCTCGCGGAGATTGACGAG – 3′ |
| 1 mut/mut–as | 5′ - TACTCGAGGTTCGGGGCAGGGTCCCCAGTCCTCGCGGAGATTCCCGAG – 3′ |
| 3-mut-I–as | 5′ - TACTCGAGACTCTAACATCCTGCAGAGT – 3′ |
| 3-mut-II–as | 5′ - TACTCGAGACTCTGGGATATTCCTGCAGAGT – 3′ |
Mutated (mut) sequences are underlined.
as, antisense; wt, wild-type.
PCR fragments were cloned into the XhoI and SacI sites of the pGL2-basic vector (Promega, Madison, Wisconsin, USA). Constructs were checked by direct sequencing using an ABI prism 310 (Applied Biosystems Perkin Elmer, Foster City, California, USA) and the primer GL1 (5′ TGT ATC TTA TGG TAC TGT AAC TG 3′).
For analysis of reporter gene expression, cells were harvested 24 hours after transfection. Luciferase activity and β-galactosidase activity (for transfection efficiency control) were measured from cellular extracts, as previously described.20,37 Luciferase activity was assessed relative to β-galactosidase activity to normalise for variations in transfection efficiency. All transfection experiments were carried out in triplicate.
Nuclear extracts and gel retardation analysis
Huh7 nuclear extracts were prepared using the modified Dignam C method.20 Gel retardation assays (electrophoretic mobility shift assay (EMSA)) were performed as previously described38 and preincubation was carried out with approximately 50 femtomol (30 000 to 50 000 cpm) 32P-labelled double stranded oligonucleotides. All oligonucleotides (table 2 ▶) were purchased from MWG Biotech.
Table 2.
Sequences of oligonucleotides used for gel retardation assays
| CREB I–s | 5′ – GAGGACTGGGGACCCTGTGACGAACATGGAG – 3′ |
| CREB I–as | 5′ – CTCCATGTTCGTCACAGGGTCCCCAGTCCTC – 3′ |
| CREB I mut–s | 5′ – GAGGACTGGGGACCCTGCCCCGAACATGGAG – 3′ |
| CREB I mut–as | 5′ – CTCCATGTTCGGGGCAGGGTCCCCAGTCCTC – 3′ |
| CREB II–s | 5′ – GCCTCTCACATCTCGTCAATCTCCGCG – 3′ |
| CREB II–as | 5′ – CGCGGAGATTGACGAGATGTGAGAGGC – 3′ |
| CREB II mut–s | 5′ – GCCTCTCACATCTCGGGAATCTCCGCG – 3′ |
| CREB II mut–as | 5′ – CGCGGAGATTCCCGAGATGTGAGAGGC – 3′ |
| Ets wt–s | 5′ – CAAACTCTGCAGGATCCCAGAGTCAG – 3′ |
| Ets wt–as | 5′ – CTGACTCTGGGATCCTGCAGAGTTTG – 3′ |
Mutated (mut) sequences are underlined.
s, sense; as, antisense; wt, wild-type.
Competition assays were carried out with increasing amounts of unlabelled oligonucleotides. The CREB wild-type consensus oligonucleotide corresponded to the sequence 5′-AGA GAT TGC CTG ACG TCA GAG AGC TAG-3′, and the CREB mutated consensus oligonucleotide to the sequence 5′′-AGA GAT TGC CTG TGG TCA GAG AGC TAG-3′.39 Supershift experiments were performed as described using antibodies against CREB1, ATF1, ATF2, ATF4, activator protein 1 (AP-1), and Ets-1/2 family members (all Santa Cruz Biotechnology).37,39 For the supershift experiments with the Ets wild-type oligonucleotide, nuclear extracts were first preincubated with cold mutated Ets oligo to reduce the unspecific signal (top arrow in fig 7C ▶).
Figure 7.
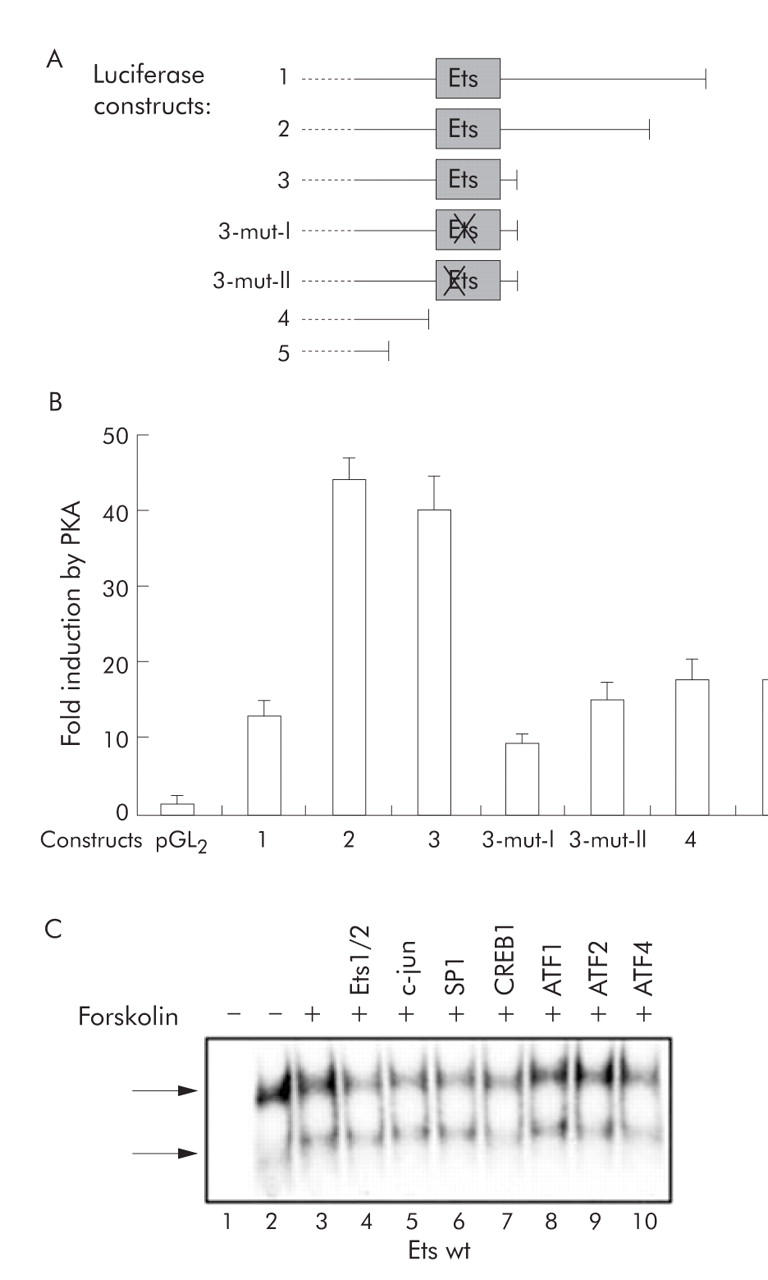
A putative Ets binding motif mediates inducible activation by protein kinase A (PKA). (A) In order to further characterise the cAMP response element binding protein (CREB)/PKA/forskolin sensitive region, the sequence of the Ets binding motif of construct 3 was mutated (constructs 3-mut-I and 3-mut-II). The constructs used for transfection experiments are shown. (B) Luciferase activity was determined in transfection experiments. Mutations of the Ets motif resulted in reduced inducibility by PKA, similar to the complete deletion of the sequence (constructs 4 and 5). (C) In electrophoretic mobility shift assay (EMSA) experiments, nuclear extracts from either unstimulated Huh7 cells (lane 2) or forskolin stimulated Huh7 cells (lanes 3–10) were incubated with a radioactive labelled double stranded oligonucleotide representing the Ets motif. In order to reduce an unspecific signal (top arrow), nuclear extracts were first preincubated with unlabelled mutated Ets oligonucleotide. Supershift EMSA analysis was performed with antibodies directed against Ets1/2 (lane 4), c-jun (lane 5), SP1 (lane 6), CREB1 (lane 7), activating transcription factor (ATF)1 (lane 8), ATF2 (lane 9), and ATF4 (lane 10). Incubation with anti-CREB1 and anti-ATF4 reduced specific complex formation (bottom arrow, lower signal).
RESULTS
PKA stimulates expression of HBV-S RNA and HBsAg
CAMP dependent signalling pathways are pivotal in regulating homeostatic functions in the liver1 and are activated during hepatic inflammation.4 We tested if CREB transcription factors influence HBV-S gene and protein expression. Replication competent HBV 1.28 wild-type plasmid was cotransfected with an expressing vector for the catalytic subunit of PKA, a strong endogenous CREB inductor. Cotransfection of HBV wild-type and 1 µg PKA in Huh7 hepatoma cells resulted in a significant increase in HBV-S-RNA in northern blot analysis 24 and 48 hours after transfection (fig 1A ▶, B). Levels of pregenomic/precore RNA were not significantly affected. After cotransfection with PKA, SHBs expression was markedly induced, as evidenced by western blot analysis 48 and 72 hours after transfection (fig 1C ▶).
Figure 1.
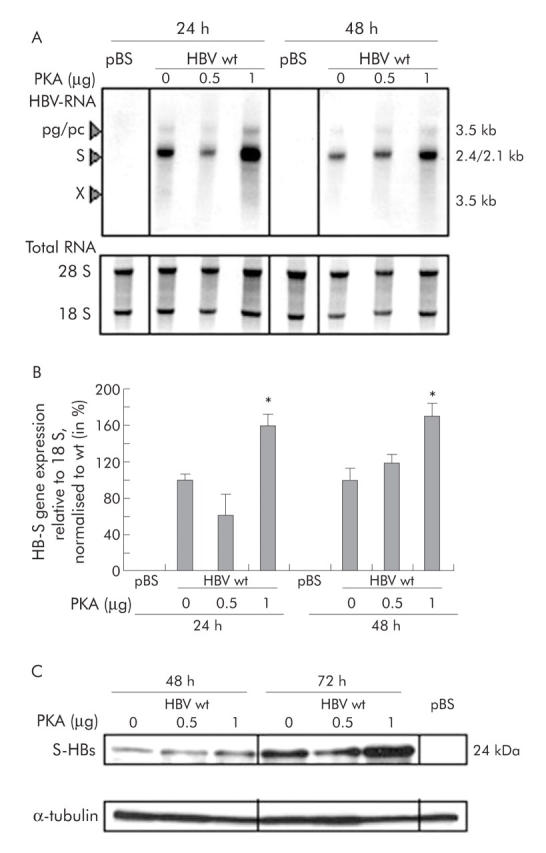
Cotransfection of hepatitis B virus (HBV) and the cAMP response element binding protein (CREB) inductor protein kinase A (PKA) leads to increased HBV-S RNA and small hepatitis B surface protein (SHB) expression. (A) Northern Blot analysis. HBV wild-type (wt) plasmid and different concentrations of PKA plasmid were cotransfected. RNA was harvested 24 and 48 hours after transfection, and HBV RNA was detected with a radioactive labelled HBV specific probe. 28S and 18S RNA signals are shown to prove equal loading of RNA. Transfection with pBluescript (pBS) alone served as a negative control. One representative northern blot experiment is shown. pg/pc, pregenomic/precore RNA. (B) Quantification of HBV-S RNA normalised to 18S. Mean (SD) based on three independent experiments. Values are given relative to PKA 0 µg. Significant difference (*p<0.05) compared with PKA 0 µg. (C) Western blot analysis. HBV wild-type (wt) plasmid and different concentrations of PKA plasmid were cotransfected. Total cellular proteins were harvested 48 and 72 hours after transfection, and S-HBs (small HBs protein) was detected with an anti-HBs antibody. Anti-α-tubulin staining of the same blot was performed to prove equal loading. Transfection with pBS served as a negative control. Cotransfection with 1 µg of PKA resulted in increased HBs protein expression.
Known pre-S2/S promoter is flanked by putative transcription factor binding sites for CREB, CEBP/β, Ets, and Stat1
The HBV pre-S2/S promoter (“S promoter”) is a TATA-less promoter and controls transcription of the 2.1 kb S-mRNA generating the middle and small surface protein. The promoter was shown to be located in the coding region of the pre-S gene between nucleotides 3045 and 3180 (EcoRI = 1) (see fig 2A ▶).8,20,40,41. To investigate the underlying mechanism of CREB/PKA dependent activation of the S promoter, we performed an initial computer analysis of the pre-S1 and pre-S2 sequences. Binding sites for transcription factors, including CREB, CEBP/β, Ets, and Stat1 were predicted downstream of the published S promoter (fig 2A ▶, B). Of note, the predicted Ets site was partially overlapping with an AP-1 motif. As it is well known that regulatory elements outside of the “core” S promoter sequence constitute part of the S promoter activity, such as upstream binding motifs for HNF3 and NF142 or a downstream Sp1 binding site,43 we then aimed to test whether these putative binding sites may contribute to S promoter activity. Therefore, a fragment covering the pre-S1/pre-S2 region, including the identified regions of interest, was amplified from a HBV wild-type plasmid and cloned into the luciferase expression vector pGL2. The resulting construct was referred to as construct 1 and is depicted in fig 2A ▶. For a detailed analysis we generated further reporter plasmids by introducing several 3′ deletions of different lengths into construct 1 (construct 1B, 2–5), as shown in fig 2A ▶.
Figure 2.
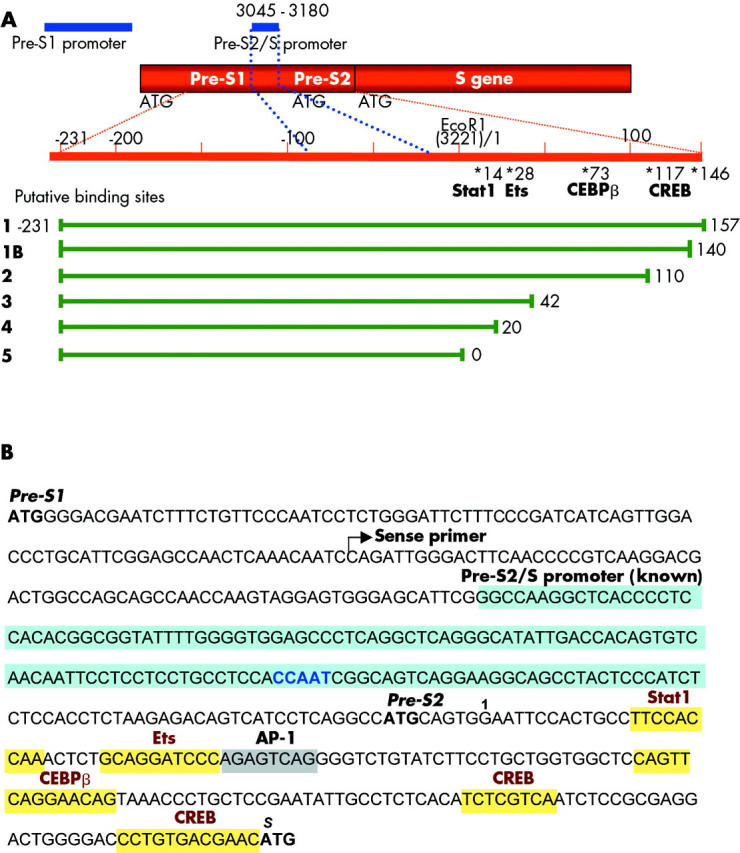
Organisation of the hepatitis B virus (HBV)-S gene, its promoters, and putative transcription factor binding sites. (A) Schematic diagram of the organisation of the HBV-S gene and its promoters (EcoR1 = 1/3221). The known region of the pre-S2/S promoter (blue) is located within the pre-S1 region from nt 3045–3180. Upstream of the S-ATG, the location of putative transcription factor binding sites for Stat1, Ets, CEBPβ, and cAMP response element binding protein (CREB) are indicated, as revealed by computer based analysis. A putative activator protein 1 (AP-1) site partially overlaps with an Ets motif. Promoter fragments of different lengths (constructs 1–5) were cloned in front of the luciferase reporter gene into the pGL2-plasmid. (B) Sequence of the pre-S1 and pre-S2 region of HBV (genotype A, subtype adw2). ATGs, the start of the applied sense primer (for luciferase constructs) and the known pre-S2/S promoter (blue) with the CCAAT binding motif are highlighted. In addition, upstream binding sites for HNF3 and NF1 plus a downstream binding site for Sp1 have been previously identified to contribute to the known pre-S2/S promoter (not shown). In the pre-S2 region, several transcription factor binding sites were predicted, and are emphasised by yellow boxes.
CREB binding motif contributes to basal S promoter activity
The reporter gene constructs (fig 2A ▶) were transfected in Huh7 human hepatoma cells, and luciferase activity was measured and normalised to β-galactosidase activity as a marker for transfection efficiency. As shown in fig 3A ▶, constructs 1 and 1B were of similar length but differed with respect to predicted CREB binding sites at positions 157 and 140 (compare with fig 2A ▶), respectively. For constructs 2–5, no CREB binding site was detected by computer analysis.
Figure 3.

cAMP response element binding protein (CREB) binding motifs contribute to basal promoter activity. (A) The full length promoter construct 1 (see fig 2A ▶) comprises two putative CREB binding motifs (CREB I/CREB II), as revealed by computer based transcription factor binding search. In addition to the 3′ deletion constructs shown in fig 2A ▶, construct 1B was generated in which the downstream CREB motif (CREB I) is deleted. (B) Luciferase constructs as shown in fig 2A ▶ and fig 3A ▶ were transfected in Huh7 cells and reporter gene expression was quantified (luciferase activity normalised to β-galactosidase activity as an indicator of transfection efficiency). Highest reporter gene expression was seen in construct 1 whereas the 3′ deletion of both putative CREB binding sites as in constructs 2–5 reduced promoter activity below 20%. Deletion of the downstream CREB site alone (construct 1B) did not alter basal promoter activity.
The highest reporter gene expression was detected in construct 1 where luciferase was under the control of the 388 bp sequence upstream from the S-ATG that includes the two putative CREB binding sites (fig 3B ▶, compare constructs 1 and 2–5). In order to determine the functional relevance of the two neighbouring putative CREB motifs in construct 1, additional 3′ deletion constructs were generated and tested for promoter activity (fig 3B ▶). Deletion of the downstream CREB site (CREB I, construct 1B) had no significant effect on basal promoter activity. In contrast, by also deleting the upstream CREB site (CREB II) as in construct 2, we observed a strong decrease in luciferase activity compared with construct 1 or 1B. These results suggest an essential role for the upstream putative CREB site (CREB II) in maintaining basal promoter activity. Further 3′ deletions as in constructs 3–5 were only correlated with a minor reduction of the basal transcriptional activity compared with construct 2, indicating that basal promoter activity is predominantly mediated by CREB II.
To assess complex formation to the CREB II motif, EMSA experiments were performed using oligonucleotides representing either the CREB II sequence, as found in the promoter or a related sequence comprising a mutation in the CREB core recognition site (fig 4A ▶). When the CREB II oligonucleotide was incubated with Huh7 nuclear extracts, complex formation was detected (fig 4B ▶, left panel). To test for specificity to CREB, competition experiments were performed using wild-type (wt) or mutated (mut) CREB consensus oligonucleotides, as described previously.39 Binding of the two slow migrating complexes could be readily reduced using low amounts of cold consensus wild-type CREB oligo, but not with mutated CREB oligo (fig 4B ▶, left panel). In contrast, incubation with a mutated CREB II oligonucleotide did not reveal specific complex formation in gelshift analysis (fig 4B ▶, right panel).
Figure 4.
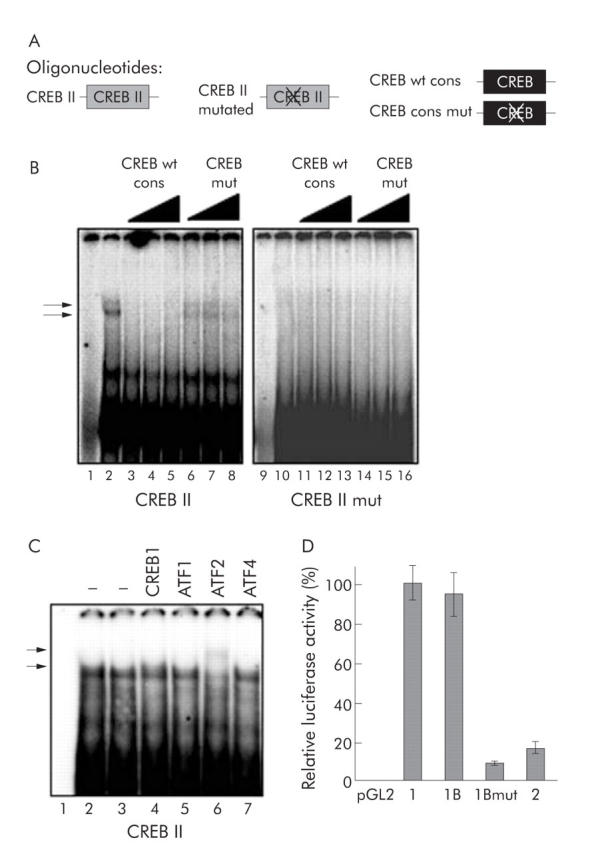
DNA binding analysis for the cAMP response element binding protein (CREB) II motif. (A) Schematic depiction of the oligonucleotides used for the EMSA experiments. CREB II oligo contains the original sequence found in the putative hepatitis B virus (HBV)-S promoter; CREB II mut has a nonsense mutation in the CREB core recognition site. Competition experiments were performed with non-labelled wild-type (wt) or mutated (mut) CREB consensus (cons) oligonucleotides. (B) In EMSA experiments, nuclear extracts from Huh7 cells (except for lanes 1 and 9) were incubated with radioactive labelled double stranded oligonucleotides corresponding to either the wild-type or a mutated sequence of the upstream CREB (CREB II) motif. In lanes 4–6 and 12–14, competition experiments with increasing concentrations of unlabelled (“cold”) consensus wild-type CREB oligo (molar ratios unlabelled:labelled 10:1, 20:1, 40:1), and in lanes 7–8 and 15–16 with cold consensus mutated CREB oligo were performed. Complex formation (arrows) was detected for the wild-type CREB II oligo, but not with the mutated CREB II oligo, that could be specifically reduced by cold CREB consensus wild-type oligonucleotides but not with mutated CREB consensus oligonucleotide. (C) In supershift EMSA experiments, nuclear extracts from Huh7 cells (except for lane 1) were incubated with radioactive labelled double stranded CREB II oligonucleotide and antibodies specific for CREB1 (lane 4), activating transcription factor (ATF) 1 (lane 5), ATF2 (lane 6), and ATF4 (lane 7). Complex formation (lower arrow) could be supershifted with ATF2 antibody (upper arrow). (D) The same mutation from the CREB II mut oligonucleotide, resulting in a lack of specific CREB complex formation, was introduced into the luciferase reporter constructs, and luciferase activity was measured after transfection in Huh7 cells, as described above. Either the mutation (construct 1B mut) or the deletion (as in construct 2) of the upstream CREB II site resulted in a tremendous decrease in luciferase activity compared with constructs 1 and 1B.
As the CREB/ATF family of transcription factors comprise a variety of different proteins able to interact with CREB binding motifs and each other,5,6 supershift experiments were carried out to define the factors involved in the binding to the CREB II motif. Complex formation with the CREB II site could be modified using an ATF2 antibody (fig 4C ▶) resulting in formation of a new slow migrating complex. These data suggest that the upstream CREB site (CREB II) mediates basal promoter activity via ATF2 binding.
To further support this finding we introduced the (nonsense) mutation represented by the “CREB II mutated” oligonucleotide into reporter construct 1B, resulting in construct 1Bmut, and performed reporter assays in Huh7 cells. As shown in fig 4D ▶, mutation of the CREB II element resulted in strong reduction of luciferase activity (to 10% of construct 1B).
Our data implied that the CREB II site mediates basal promoter activity via ATF2 binding whereas the CREB I motif appeared to be dispensable for transcriptional activation. However, due to the close proximity of both elements, functional compensation of CREB II by CREB I could not be excluded. In EMSA experiments, CREB I showed the same CREB specific complex formation as seen for CREB-II (compare with fig 4B ▶, data not shown). To test for a potential functional redundancy, we analysed the CREB I element for specific binding of CREB/ATF factors by supershift EMSA according to the experiment shown in fig 4C ▶. Interestingly, we detected specific binding of the transcription factors CREB1 and ATF2 to the CREB I site (fig 5A ▶), thereby showing a similar binding pattern as for the CREB II element. In order to examine whether this binding affinity to CREB I corresponds to a compensatory function for CREB II in the promoter, we introduced mutations into CREB I and/or CREB II core sequences of construct 1 (fig 5B ▶).
Figure 5.
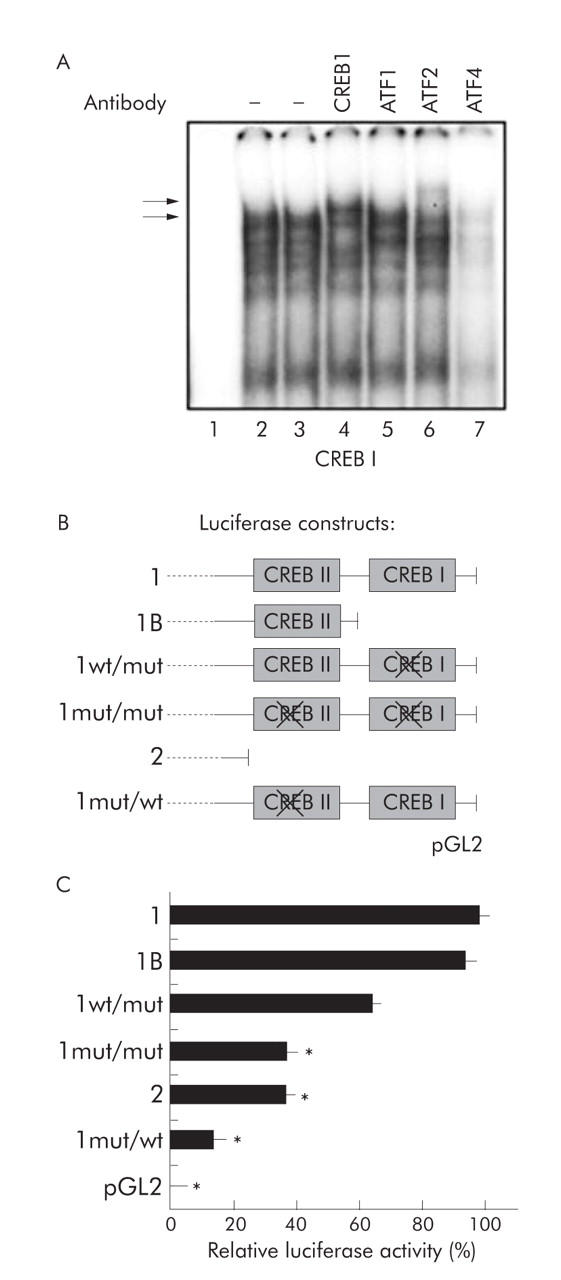
Analysis of neighbouring cAMP response element binding protein (CREB) sites for DNA binding specificity and functional redundancy. (A) The neighbouring downstream CREB I site was tested for complex formation in supershift EMSA experiments (as shown in fig 4C ▶ for CREB II). Nuclear extracts from Huh7 cells (except for lane 1) were incubated with radioactive labelled double stranded CREB I oligonucleotide and antibodies specific for CREB1 (lane 4), activating transcription factor (ATF)1 (lane 5), ATF2 (lane 6), and ATF4 (lane 7). Complex formation (lower arrow) could be supershifted with ATF2 and CREB1 antibodies (upper arrow). (B) To determine if the two CREB sites can compensate for each other’s loss of function, additional mutated luciferase constructs were generated as schematically shown bearing nonsense mutations either in one or both CREB motifs. (C) Relative luciferase activity was assessed in transfection experiments. Nonsense mutation of the downstream CREB site (CREB I, construct 1wt/mut) as well as deletion of this motif (construct 1B) resulted only in minor reduction of basal promoter activity whereas deletion (construct 2) or mutation of the upstream CREB site (CREB II, construct 1mut/wt) strongly decreased relative luciferase expression (*p<0.05 compared with construct 1). Introducing mutations of both CREB sites into the full length construct (construct 1mut/mut) had a similar effect.
The reporter plasmids were transfected into Huh7 cells and luciferase activity was determined and compared with controls comprising either both CREB sites (construct 1), one (construct 1B), or no CREB sites (construct 2). Of note, we did not observe any compensatory function of the CREB I site. Instead, a reporter plasmid containing a mutated CREB II element and a functional CREB I site (1mut/wt) showed only approximately 15% promoter activity compared with wild-type construct 1.
S promoter activity is inducible via CREB, PKA, and forskolin
Our analysis identified CREB binding motifs in the pre-S2 sequence contributing to basal S promoter activity. Thus we next investigated whether cAMP signalling pathways also mediate inducible S promoter activation, as indicated by the cotransfection experiment of HBV wild-type and the PKA expression plasmid (fig 1 ▶). The reporter gene constructs 1–5 (see fig 2A ▶) were cotransfected with CREB and/or PKA expressing plasmids.39 As expected from the previous results, construct 1 had the highest basal activity but also showed inducibility by CREB and PKA (fig 6A ▶). Constructs 2–5 had low basal luciferase expression but luciferase activity was strongly induced by CREB and even further enhanced by PKA in constructs 2 and 3. Constructs 4 and 5 also displayed some inducible activity but their relative response to CREB or PKA was remarkably reduced compared with constructs 2 or 3. This suggests that a significant part of the CREB/PKA inducible activity is mediated by the sequence distinguishing constructs 3 and 4, and sequence analysis revealed that constructs 1–3 carry a putative Ets binding motif which was deleted in constructs 4 and 5.
Figure 6.
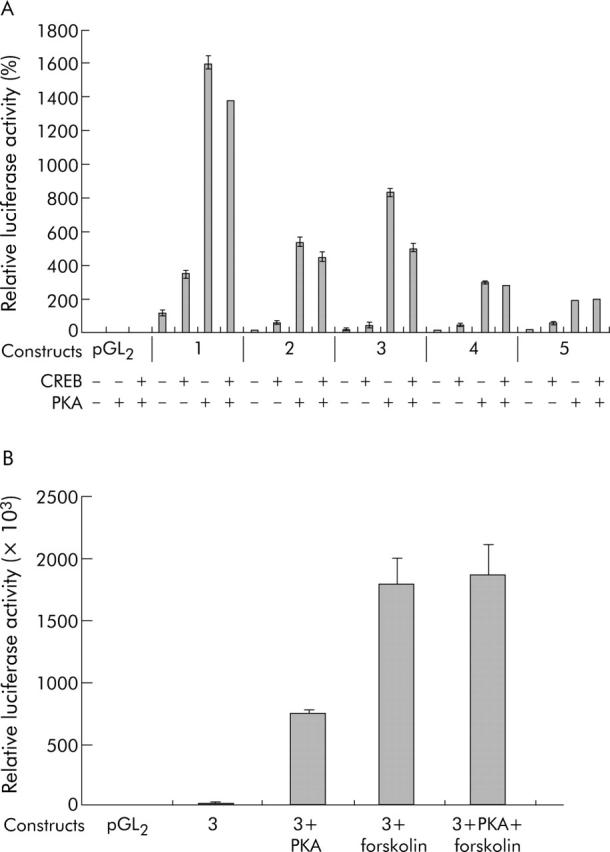
Identification of a region conferring inducible activity via cAMP response element binding protein (CREB), protein kinase A (PKA), and the exogenous PKA activator forskolin. (A) Deletion constructs 1–5 (see fig 2A ▶) were cotransfected in Huh7 hepatoma cells with 50 ng CREB-plasmid and/or 1 µg PKA expression plasmid, as indicated. Construct 1 conferred the highest basal activity (as expected from figs 3 ▶–5 ▶). Luciferase expression was highly induced by cotransfection of CREB and even further enhanced by PKA in all constructs. However, the highest relative induction was seen in constructs 2 and 3 whereas this inducible activity was remarkably lower in constructs 4 and 5. Constructs 1–3 contain a putative Ets-1 binding motif, which was not present in constructs 4 and 5. (B) Forskolin is a chemical inductor of PKA/CREB signalling pathways. Construct 3 was either cotransfected with PKA and/or cells were stimulated with 10 µM forskolin, as indicated. Stimulation with forskolin resulted in maximal activation of relative luciferase expression.
Forskolin is a chemical inductor of PKA/CREB signalling pathways. It increases endogenous PKA transcription and subsequently activates CREB.39,44 In order to test if extracellular signals or exogenous activators may also enhance S promoter activity, construct 3 was either cotransfected with PKA and/or cells were stimulated with 10 µM forskolin. Stimulation with forskolin resulted in maximal activation of relative luciferase expression that could not be further stimulated by cotransfecting the PKA expressing construct (fig 6B ▶).
These experiments indicated that the putative Ets binding motif might be involved in mediating PKA dependent promoter activation. We thus mutated the putative Ets sequences in this part of the promoter region (constructs 3-mut-I and 3-mut-II, fig 7A ▶). The mutant Ets constructs were then cotransfected with the catalytic subunit of PKA. These experiments showed that the intact Ets motif was necessary to confer full PKA mediated gene activation of the S promoter (fig 7B ▶).
We next attempted to further characterise possible transcription factors involved in binding to the Ets motif. The family of Ets transcription factors consists of more than 40 different proteins, and protein-protein interactions (for example, between AP1 and Ets) are common.45,46 We therefore tested a variety of different factor specific antibodies in supershift analysis. Nuclear extracts from either unstimulated or forskolin stimulated Huh7 cells were incubated with radioactive labelled double stranded Ets or mutated Ets oligonucleotides in EMSA experiments. Only for the Ets wild-type oligo did forskolin stimulation result in formation of a novel complex (fig 7C ▶, bottom arrow, and data not shown). Supershift experiments were performed with antibodies directed against Ets1/2, c-jun, SP1, CREB1, ATF1, ATF2, and ATF4. These experiments showed a reduction in complex formation of the slower migrating forskolin inducible signal (fig 7C ▶, bottom arrow) when extracts were incubated with antibodies directed against CREB1 and ATF4 (fig 7C ▶). This indicates that the complex formed after forskolin stimulation with the “Ets-wt” oligonucleotide contains both CREB1 and ATF4 proteins.
DISCUSSION
Transcriptional regulation via adenylate cyclase signalling pathways is mediated by a family of cyclic AMP responsive nuclear factors, including CREB. The activation function of CRE binding proteins is modulated by phosphorylation by several kinases (for example, PKA) and other cellular cofactors.6 CREB transcription factors play a pivotal role in the liver as they are essential for regulating hepatic metabolic functions, such as gluconeogenesis or lipid oxidation, as well as controlling hepatocyte proliferation.1–3 In addition, CRE pathways are activated in experimental settings of hepatic inflammation via increasing c-jun.4 In this study, we identified a role for CREB/PKA sensitive signalling in maintaining basal promoter activity and inducible expression of the hepatitis B virus pre-S2/S promoter.
Regulation of HBV-S gene expression is crucial for viral pathogenesis, as the envelope proteins are essential for virus formation, infectivity, and immunogenicity. The strongest regulatory element within the HBV genome is the pre-S2/S promoter (“S promoter”), located within the pre-S region, subsequently causing the release of HBs particles.7,29 The extraordinary high prevalence of naturally occurring pre-S mutants during chronic HBV infection have led to identification of several pre-S mutations affecting the S promoter, thereby providing a molecular explanation for an unfavourable clinical outcome seen in many of these patient populations.9–16 The highest reduction in HBsAg expression can be detected when the CCAAT motif in the HBV pre-S2/S promoter is affected, although even introduction of nonsense mutations into the CCAAT motif into promoter constructs with otherwise normal sequence shows a residual promoter activity of approximately 25%,20 thereby suggesting alternative signalling pathways for maintaining basal viral promoter activity.
As CREB/PKA pathways are central in hepatocyte signal transduction, we tested whether HBV-S gene and envelope protein expression are upregulated on CREB/PKA activation. In fact, cotransfection of HBV wild-type and PKA expression plasmid in hepatoma cells resulted in increased HBV-S RNA and SHBs protein expression (fig 1 ▶). By applying reporter gene and DNA binding analysis experiments, we identified two novel regulatory elements in the pre-S2 sequence, possibly mediating this CREB/PKA sensitive S promoter activation—a CREB transcription factor binding motif in the pre-S2 region contributing to basal S promoter activity via binding of ATF2, and a predicted Ets site in the pre-S2 region mediating inducible activation of gene expression by PKA/forskolin, possibly involving CREB1 and ATF4. Interestingly, these regulatory regions are located within the transcribed (pre-S2) region of the HBV genome. Given the dense organisation of the circular HBV DNA genome, the contribution of regulatory elements within the transcribed region to the activity of the previously described upstream “core” S promoter is not unexpected. Similar promoter organisations with regulatory elements located in intronic regions have been shown for a variety of promoters, such as CD95 or caspase 8.37,47
Our results showed that the basal activity of the HBV pre-S2/S promoter is supported by a CREB dependent mechanism (figs 3 ▶–5 ▶). We identified two putative CREB motifs with similar binding patterns in the pre-S2 region, and supershift analysis suggested that ATF2 is involved in complex formation at these CREB binding motifs. Although the DNA binding assays implied a functional redundancy for both CREB sites, only the upstream CREB site appeared essential for full basal promoter activity in our reporter gene experiments. Additional observations indicate the functional relevance of these CREB sites. In order to characterise pre-S2 protein function, Le Seyec et al generated HBV constructs with deletions in the pre-S2 region. They observed that deletion of any of the two CREB motifs (deletion L144/153 = “CREB-II”, deletion L154/163 = “CREB-I”) was associated with a reduction in HBsAg secretion of between 15% and 30% in vitro.19 Interestingly, deletion of the downstream CREB motif in HBV constructs was associated with HBsAg reduction. This would argue that the downstream CREB site may functionally compensate for loss of the upstream site in HBV deletion constructs, although this was not observed in our experiments applying the luciferase reporter system.
We also observed inducible upregulation of promoter activity by CREB and its activators PKA or forskolin (figs 6 ▶, 7 ▶). Our reporter gene experiments showed that a sequence in the pre-S2 region comprising an Ets motif was significantly involved in mediating cAMP dependent activation. The family of Ets transcription factors consists of more than 40 different proteins, and protein-protein interactions (for example, between AP1 and Ets) are common.45,46 Our DNA binding experiments pointed towards involvement of Ets factors and/or AP-1 as forskolin/PKA inducible complex formation could be detected towards these elements. However, supershift experiments with anti-CREB1 and anti-ATF4 antibodies reduced complex formation, indicating inducible heterodimer binding of two CREB family members to this element. Earlier results showed that ATF-1/2/4 can form dimers with other proteins (for example, c-jun) which then activate the AP-1 transcription factor complex.48 AP-1 sites are often closely located to Ets sites—as also seen in the putative S promoter (see fig 2 ▶)—and can interact with Ets binding.45,46 Our supershift experiments suggested that CREB1 and ATF4 are involved in complex formation, thereby potentially activating transcription via the Ets motif. However, we cannot exclude the fact that more than the identified factors are involved in this process.
Taken together, our experiments show that CREB motifs in the pre-S2 region contribute to basal S promoter activity and that CREB/PKA can upregulate HBsAg expression. However, the true in vivo relevance of these in vitro findings needs to be further addressed (for example, by coinjecting HBV and PKA plasmids in a mouse model of acute HBV infection and monitoring HBsAg expression49 or by assessing HBsAg levels after acute infection in CREB knockout mice).2 Our experiments indicate a novel mechanism by which HBV is able to utilise the cellular signal transduction machinery of hepatocytes in order to enhance its HBsAg expression level.
Acknowledgments
This work was supported by Hep-Net, the German network of competence for viral hepatitis, the EU-grant of Virgil, and the German Research Foundation (TA 434/1-1 to FT). The authors are grateful to Tom Luedde and Nadine Gröger for valuable discussion and technical assistance.
Abbreviations
HBV, hepatitis B virus
CREB, cAMP response element binding protein
PKA, protein kinase A
cAMP, cyclic adenosine monophosphate
HBsAg, hepatitis B surface antigen
SHBs/MHBs/LHBs, small/middle/large hepatitis B surface protein
EMSA, electrophoretic mobility shift assay
ATF, activating transcription factor
ORF, open reading frame
pBS, Bluescript plasmid
AP-1, activator protein 1
Published online first 6 May 2005
Conflict of interest: None declared.
REFERENCES
- 1.Servillo G, Della Fazia MA, Sassone-Corsi P. Coupling cAMP signaling to transcription in the liver: pivotal role of CREB and CREM. Exp Cell Res 2002;275:143–54. [DOI] [PubMed] [Google Scholar]
- 2.Herzig S, Hedrick S, Morantte I, et al. CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 2003;426:190–3. [DOI] [PubMed] [Google Scholar]
- 3.Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature 2001;413:179–83. [DOI] [PubMed] [Google Scholar]
- 4.Zhang B, Liu S, Perpetua MD, et al. Cytokines increase CRE binding but decrease CRE-mediated reporter activity in rat hepatocytes by increasing c-Jun. Hepatology 2004;39:1343–52. [DOI] [PubMed] [Google Scholar]
- 5.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol 2001;2:599–609. [DOI] [PubMed] [Google Scholar]
- 6.Papavassiliou AG. The CREB/ATF family of transcription factors: modulation by reversible phosphorylation. Anticancer Res 1994;14:1801–5. [PubMed] [Google Scholar]
- 7.Tacke F, Manns MP, Trautwein C. Influence of mutations in the hepatitis B virus genome on virus replication and drug resistance—implications for novel antiviral strategies. Curr Med Chem 2004;11:2667–77. [DOI] [PubMed] [Google Scholar]
- 8.Locarnini S, McMillan J, Bartholomeusz A. The hepatitis B virus and common mutants. Semin Liver Dis 2003;23:5–20. [DOI] [PubMed] [Google Scholar]
- 9.Bock CT, Tillmann HL, Maschek HJ, et al. A preS mutation isolated from a patient with chronic hepatitis B infection leads to virus retention and misassembly. Gastroenterology 1997;113:1976–82. [DOI] [PubMed] [Google Scholar]
- 10.Trautwein C, Schrem H, Tillmann HL, et al. Hepatitis B virus mutations in the pre-S genome before and after liver transplantation. Hepatology 1996;24:482–8. [DOI] [PubMed] [Google Scholar]
- 11.Preikschat P, Gunther S, Reinhold S, et al. Complex HBV populations with mutations in core promoter, C gene, and pre-S region are associated with development of cirrhosis in long-term renal transplant recipients. Hepatology 2002;35:466–77. [DOI] [PubMed] [Google Scholar]
- 12.Fan YF, Lu CC, Chen WC, et al. Prevalence and significance of hepatitis B virus (HBV) pre-S mutants in serum and liver at different replicative stages of chronic HBV infection. Hepatology 2001;33:277–86. [DOI] [PubMed] [Google Scholar]
- 13.Carman WF, Trautwein C, van Deursen FJ, et al. Hepatitis B virus envelope variation after transplantation with and without hepatitis B immune globulin prophylaxis. Hepatology 1996;24:489–93. [DOI] [PubMed] [Google Scholar]
- 14.Kalinina T, Riu A, Fischer L, et al. A dominant hepatitis B virus population defective in virus secretion because of several S-gene mutations from a patient with fulminant hepatitis. Hepatology 2001;34:385–94. [DOI] [PubMed] [Google Scholar]
- 15.Sugauchi F, Ohno T, Orito E, et al. Influence of hepatitis B virus genotypes on the development of preS deletions and advanced liver disease. J Med Virol 2003;70:537–44. [DOI] [PubMed] [Google Scholar]
- 16.Raimondo G, Costantino L, Caccamo G, et al. Non-sequencing molecular approaches to identify preS2-defective hepatitis B virus variants proved to be associated with severe liver diseases. J Hepatol 2004;40:515–9. [DOI] [PubMed] [Google Scholar]
- 17.Bock CT, Tillmann HL, Manns MP, et al. The pre-S region determines the intracellular localization and appearance of hepatitis B virus. Hepatology 1999;30:517–25. [DOI] [PubMed] [Google Scholar]
- 18.Le Seyec J, Chouteau P, Cannie I, et al. Infection process of the hepatitis B virus depends on the presence of a defined sequence in the pre-S1 domain. J Virol 1999;73:2052–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Seyec J, Chouteau P, Cannie I, et al. Role of the pre-S2 domain of the large envelope protein in hepatitis B virus assembly and infectivity. J Virol 1998;72:5573–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bock CT, Kubicka S, Manns MP, et al. Two control elements in the hepatitis B virus S-promoter are important for full promoter activity mediated by CCAAT-binding factor. Hepatology 1999;29:1236–47. [DOI] [PubMed] [Google Scholar]
- 21.Raney AK. Transcriptional control of hepatitis B virus. Methods Mol Med 2004;95:303–14. [DOI] [PubMed] [Google Scholar]
- 22.Raney AK, Le HB, McLachlan A. Regulation of transcription from the hepatitis B virus major surface antigen promoter by the Sp1 transcription factor. J Virol 1992;66:6912–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raney AK, Milich DR, McLachlan A. Complex regulation of transcription from the hepatitis B virus major surface antigen promoter in human hepatoma cell lines. J Virol 1991;65:4805–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaul Y, Ben Levy R, De Medina T. High affinity binding site for nuclear factor I next to the hepatitis B virus S gene promoter. EMBO J 1986;5:1967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neurath AR, Kent SB, Strick N, et al. Identification and chemical synthesis of a host cell receptor binding site on hepatitis B virus. Cell 1986;46:429–36. [DOI] [PubMed] [Google Scholar]
- 26.Cooper A, Paran N, Shaul Y. The earliest steps in hepatitis B virus infection. Biochim Biophys Acta 2003;1614:89–96. [DOI] [PubMed] [Google Scholar]
- 27.Fernholz D, Galle PR, Stemler M, et al. Infectious hepatitis B virus variant defective in pre-S2 protein expression in a chronic carrier. Virology 1993;194:137–48. [DOI] [PubMed] [Google Scholar]
- 28.Santantonio T, Jung MC, Schneider R, et al. Hepatitis B virus genomes that cannot synthesize pre-S2 proteins occur frequently and as dominant virus populations in chronic carriers in Italy. Virology 1992;188:948–52. [DOI] [PubMed] [Google Scholar]
- 29.Seeger C, Mason WS. Hepatitis B virus biology. Microbiol Mol Biol Rev 2000;64:51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee WM. Hepatitis B virus infection. N Engl J Med 1997;337:1733–45. [DOI] [PubMed] [Google Scholar]
- 31.Bock CT, Tillmann HL, Torresi J, et al. Selection of hepatitis B virus polymerase mutants with enhanced replication by lamivudine treatment after liver transplantation. Gastroenterology 2002;122:264–73. [DOI] [PubMed] [Google Scholar]
- 32.Tacke F, Trautwein C, Zhao S, et al. Quantification of hepatic thrombopoietin mRNA transcripts in patients with chronic liver diseases shows maintained gene expression in different etiologies of liver cirrhosis. Liver 2002;22:205–12. [DOI] [PubMed] [Google Scholar]
- 33.Tacke F, Gehrke C, Luedde T, et al. Basal core promoter and precore mutations in the hepatitis B virus genome enhance replication efficacy of lamivudine-resistant mutants. J Virol 2004;78:8524–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tacke F, Marini FC III, Zhao S, et al. Expression of inducible Bcl-X(S) in myeloid leukemia: compensatory upregulation of Bcl-X(L) and Bcl-2 prevents apoptosis and chemosensitization. Cancer Biol Ther 2004;3:340–7. [DOI] [PubMed] [Google Scholar]
- 35.Tacke F, Schoffski P, Luedde T, et al. Analysis of factors contributing to higher erythropoietin levels in patients with chronic liver disease. Scand J Gastroenterol 2004;39:259–66. [DOI] [PubMed] [Google Scholar]
- 36.Bock CT, Buerke B, Tillmann HL, et al. Relevance of hepatitis B core gene deletions in patients after kidney transplantation. Gastroenterology 2003;124:1809–20. [DOI] [PubMed] [Google Scholar]
- 37.Liedtke C, Groger N, Manns MP, et al. The human caspase-8 promoter sustains basal activity through SP1 and ETS-like transcription factors and can be up-regulated by a p53-dependent mechanism. J Biol Chem 2003;278:27593–604. [DOI] [PubMed] [Google Scholar]
- 38.Ludde T, Kubicka S, Plumpe J, et al. Ras adenoviruses modulate cyclin E protein expression and DNA synthesis after partial hepatectomy. Oncogene 2001;20:5264–78. [DOI] [PubMed] [Google Scholar]
- 39.Niehof M, Manns MP, Trautwein C. CREB controls LAP/C/EBP beta transcription. Mol Cell Biol 1997;17:3600–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Antonucci TK, Rutter WJ. Hepatitis B virus (HBV) promoters are regulated by the HBV enhancer in a tissue-specific manner. J Virol 1989;63:579–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chang HK, Ting LP. The surface gene promoter of the human hepatitis B virus displays a preference for differentiated hepatocytes. Virology 1989;170:176–83. [DOI] [PubMed] [Google Scholar]
- 42.Raney AK, McLachlan A. Characterization of the hepatitis B virus major surface antigen promoter hepatocyte nuclear factor 3 binding site. J Gen Virol 1997;78:3029–38. [DOI] [PubMed] [Google Scholar]
- 43.Raney AK, McLachlan A. Characterization of the hepatitis B virus large surface antigen promoter Sp1 binding site. Virology 1995;208:399–404. [DOI] [PubMed] [Google Scholar]
- 44.Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 1989;59:675–80. [DOI] [PubMed] [Google Scholar]
- 45.Li R, Pei H, Watson DK. Regulation of Ets function by protein-protein interactions. Oncogene 2000;19:6514–23. [DOI] [PubMed] [Google Scholar]
- 46.Macleod K, Leprince D, Stehelin D. The ets gene family. Trends Biochem Sci 1992;17:251–6. [DOI] [PubMed] [Google Scholar]
- 47.Muller M, Wilder S, Bannasch D, et al. p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J Exp Med 1998;188:2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin WC, Shen BJ, Tsay YG, et al. Transcriptional activation of C/EBPbeta gene by c-Jun and ATF2. DNA Cell Biol 2002;21:551–60. [DOI] [PubMed] [Google Scholar]
- 49.Klein C, Bock CT, Wedemeyer H, et al. Inhibition of hepatitis B virus replication in vivo by nucleoside analogues and siRNA. Gastroenterology 2003;125:9–18. [DOI] [PubMed] [Google Scholar]